(tucatinib)
14 CLINICAL STUDIES
14 CLINICAL STUDIES
14.1 HER2-Positive Metastatic Breast Cancer
The efficacy of TUKYSA in combination with trastuzumab and capecitabine was evaluated in 612 patients in HER2CLIMB (NCT02614794), a randomized (2:1), double-blind, placebo-controlled trial. Patients were required to have HER2-positive, unresectable locally advanced or metastatic breast cancer, with or without brain metastases, and prior treatment with trastuzumab, pertuzumab, and ado-trastuzumab emtansine (T-DM1) separately or in combination, in the neoadjuvant, adjuvant or metastatic setting. HER2 positivity was based on archival or fresh tissue tested with an FDA-approved test at a central laboratory prior to enrollment with HER2 positivity defined as HER2 IHC 3+ or ISH positive.
Patients with brain metastases, including those with progressing or untreated lesions, were eligible provided they were neurologically stable and did not require immediate radiation or surgery. The trial excluded patients with leptomeningeal disease. Randomization was stratified by the presence or history of brain metastases (yes vs. no), Eastern Cooperative Oncology Group (ECOG) performance status (0 vs. 1), and region (U.S., Canada, or rest of world).
Patients received TUKYSA 300 mg or placebo orally twice daily with a trastuzumab or a non-US approved trastuzumab product loading dose of 8 mg/kg on Day 1 of Cycle 1 if needed and then a maintenance dose of 6 mg/kg on Day 1 of every 21-day cycle thereafter and capecitabine 1000 mg/m2 orally twice daily on Days 1 through 14 of every 21-day cycle. An alternate trastuzumab dosing regimen was 600 mg administered subcutaneously on Day 1 of every 21-day cycle. Patients were treated until disease progression or unacceptable toxicity. Tumor assessments, including brain-MRI in patients with presence or history of brain metastases at baseline, occurred every 6 weeks for the first 24 weeks and every 9 weeks thereafter.
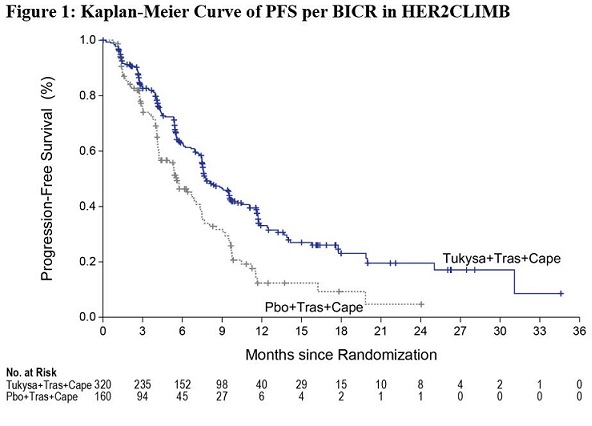
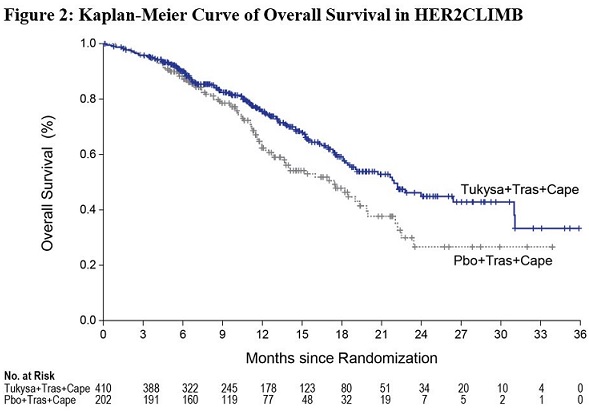
The major efficacy outcome measure was progression-free survival (PFS) in the first 480 randomized patients assessed by blinded independent central review (BICR) using Response Evaluation Criteria in Solid Tumors (RECIST) v1.1. Additional efficacy outcome measures were evaluated in all randomized patients and included overall survival (OS), PFS among patients with a history or presence of brain metastases (PFSBrainMets), and confirmed objective response rate (ORR).
The median age was 54 years (range: 22 - 82); 116 (19%) patients were age 65 or older. The majority were White (73%) and female (99%) and 51% had an ECOG performance status of 1. Sixty percent had estrogen and/or progesterone receptor-positive disease. Forty-eight percent had a presence or history of brain metastases; of these patients, 23% had untreated brain metastases, 40% had treated but stable brain metastases, and 37% had treated but radiographically progressing brain metastases. Seventy-four percent of patients had visceral metastases. Patients had received a median of 4 (range, 2 to 17) prior lines of systemic therapy and a median of 3 (range, 1 to 14) prior lines of systemic therapy in the metastatic setting. All patients received prior trastuzumab and T-DM1 and all but two patients had prior pertuzumab.
Efficacy results are summarized in Table 12 and Figure 1 and 2. Efficacy results were consistent across patient subgroups defined by stratification factors (presence or history of brain metastases, ECOG status, region of world) and hormone receptor status.
| BICR=blinded independent central review; CI=confidence interval; PFS=progression-free survival; OS=overall survival; ORR=objective response rate; CR=complete response; PR=partial response; DOR=duration of response. | ||
| ||
TUKYSA + Trastuzumab + Capecitabine | Placebo + Trastuzumab + Capecitabine | |
PFS* | N=320 | N=160 |
Number of events (%) | 178 (56) | 97 (61) |
Median, months (95% CI) | 7.8 (7.5, 9.6) | 5.6 (4.2, 7.1) |
Hazard ratio (95% CI)† | 0.54 (0.42, 0.71) | |
P-value‡ | <0.00001 | |
OS | N=410 | N=202 |
Number of deaths (%) | 130 (32) | 85 (42) |
Median, months (95% CI) | 21.9 (18.3, 31.0) | 17.4 (13.6, 19.9) |
Hazard ratio (95% CI)† | 0.66 (0.50, 0.87) | |
P-value§ | 0.00480 | |
PFSBrainMets¶ | N=198 | N=93 |
Number of events (%) | 106 (53.5) | 51 (54.8) |
Median, months (95% CI) | 7.6 (6.2, 9.5) | 5.4 (4.1, 5.7) |
Hazard ratio (95% CI)† | 0.48 (0.34, 0.69) | |
P-value# | <0.00001 | |
Confirmed ORR for Patients with Measurable Disease | N=340 | N=171 |
ORR (95% CI)Þ | 40.6 (35.3, 46.0) | 22.8 (16.7, 29.8) |
CR (%) | 3 (0.9) | 2 (1.2) |
PR (%) | 135 (39.7) | 37 (21.6) |
P-value‡ | 0.00008 | |
DOR | ||
Median, months (95% CI)ß | 8.3 (6.2, 9.7) | 6.3 (5.8, 8.9) |
14.2 HER2-Positive Metastatic Colorectal Cancer
The efficacy of TUKYSA in combination with trastuzumab was evaluated in 84 patients in MOUNTAINEER (NCT03043313), an open-label, multicenter trial. Patients were required to have HER2-positive, RAS wild-type, unresectable or metastatic colorectal cancer and prior treatment with fluoropyrimidines, oxaliplatin, irinotecan, and anti-vascular endothelial growth factor (VEGF) monoclonal antibody (mAb). Patients whose disease had deficient mismatch repair (dMMR) proteins or microsatellite instability-high (MSI-H) must have also received an anti-programmed cell death protein-1 (PD-1) mAb. Patients who received prior anti-HER2 targeting therapy were excluded. HER2 positivity as defined by HER2 overexpression or gene amplification was prospectively determined in local laboratories using immunohistochemistry (IHC), in situ hybridization (ISH), and/or next generation sequencing (NGS) on tumor tissue. RAS status was performed as standard of care prior to study entry based on expanded RAS testing including KRAS exons 2, 3, and 4 and NRAS exons 2, 3, and 4.
The median age was 55 years (range: 24 to 77); 12 (14%) patients were age 65 or older. Seventy-seven percent of patients were White, 4% were Black, 4% were Asian, and 4% were Hispanic or Latino. Sixty-one percent of patients were male. Seventy percent of patients had lung metastases, 64% had liver metastases, 60% had an ECOG performance status of 0, 37% had an ECOG performance status of 1, and 4% had an ECOG performance status of 2. Ninety-nine percent of patients received prior treatment with fluoropyrimidine, oxaliplatin, and irinotecan and 83% and 52% received anti-VEGF antibodies and anti-EGFR antibodies respectively; 23%, 38%, and 39% received 1, 2, or ≥3 prior lines of therapy, respectively.
Patients received TUKYSA 300 mg orally twice per day with a loading dose of trastuzumab or a non-US approved trastuzumab product 8 mg/kg intravenously on Day 1 of Cycle 1, followed by a maintenance dose of trastuzumab 6 mg/kg on Day 1 of each subsequent 21-day cycle. Patients were treated until disease progression or unacceptable toxicity.
The major efficacy outcome measures were overall response rate (ORR) and duration of response (DOR) as assessed by blinded independent central review (BICR) according to RECIST version 1.1.
Efficacy results are summarized in Table 13.
TUKYSA + trastuzumab | |
Overall Response Rate (%) (95% CI) | 38 (28, 49) |
CR (%) | 3 (3.6) |
PR (%) | 29 (35) |
Duration of Response | N=32 |
Median*, months (95% CI) | 12.4 (8.5, 20.5) |
Patients with DOR≥6 months† | 81% |
Patients with DOR≥12 months† | 34% |
MEDICATION GUIDE
| This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 01/2023 | |||
PATIENT INFORMATION | |||
Important information: If your healthcare provider prescribes TUKYSA in combination with capecitabine for your breast cancer, also read the Patient Information that comes with capecitabine. | |||
What is TUKYSA?
It is not known if TUKYSA is safe and effective in children. | |||
Before taking TUKYSA, tell your healthcare provider about all of your medical conditions, including if you: | |||
| |||
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. TUKYSA may affect the way your other medicines work, and other medicines may affect the way TUKYSA works. Know the medicines you take. Keep a list of all the medicines you take and show it to your healthcare provider and pharmacist every time you get a new medicine. | |||
How should I take TUKYSA? | |||
| |||
What are the possible side effects of TUKYSA? | |||
| |||
|
| ||
The most common side effects of TUKYSA in combination with trastuzumab and capecitabine in adults with HER2-positive breast cancer include: | |||
|
| ||
The most common side effects of TUKYSA in combination with trastuzumab in adults with RAS wild-type HER2-positive colorectal cancer include: | |||
|
| ||
Your healthcare provider may change your dose of TUKYSA, temporarily stop, or permanently stop treatment with TUKYSA if you have certain side effects. | |||
How should I store TUKYSA? | |||
| |||
Keep TUKYSA and all medicines out of reach of children. | |||
General information about the safe and effective use of TUKYSA. | |||
What are the ingredients in TUKYSA? Tablet coating: yellow film coat: polyvinyl alcohol, titanium dioxide, macrogol/polyethylene glycol, talc, and yellow iron oxide non-irradiated. | |||
Additional Resources
Chat online with Pfizer Medical Information regarding your inquiry on a Pfizer medicine or vaccine.
Speak with a Pfizer Medical Information Professional regarding your Pfizer medicine or vaccine inquiry.
Available 9AM-5PM ET Monday to Friday; excluding holidays.
Submit a medical question for a Pfizer medicine or a vaccine.
The submission will be reviewed during our standard business hours.
To report an adverse event related to a Pfizer product and you are not part of a clinical trial* for this medication, click the link below to submit your information:
Pfizer Safety Reporting Site
*If you are involved in a clinical trial for either product, adverse events should be reported to your coordinating study site.
If you cannot use the above website to report an adverse event related to a Pfizer medication, please call (800) 438-1985.
You may also contact the U.S. Food and Drug Administration (FDA) directly to report adverse events or product quality concerns either online at www.fda.gov/medwatch or by calling (800) 332-1088.