(trastuzumab-qyyp)
6 ADVERSE REACTIONS
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the label:
- •
- Cardiomyopathy [see Warnings and Precautions (5.1)]
- •
- Infusion Reactions [see Warnings and Precautions (5.2)]
- •
- Embryo-Fetal Toxicity [see Warnings and Precautions (5.3)]
- •
- Pulmonary Toxicity [see Warnings and Precautions (5.4)]
- •
- Exacerbation of Chemotherapy-Induced Neutropenia [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The most common adverse reactions in patients receiving trastuzumab products in the adjuvant and metastatic breast cancer setting are fever, nausea, vomiting, infusion reactions, diarrhea, infections, increased cough, headache, fatigue, dyspnea, rash, neutropenia, anemia, and myalgia. Adverse reactions requiring interruption or discontinuation of trastuzumab product treatment include CHF, significant decline in left ventricular cardiac function, severe infusion reactions, and pulmonary toxicity [see Dosage and Administration (2.5)].
In the metastatic gastric cancer setting, the most common adverse reactions (≥ 10%) that were increased (≥ 5% difference) in the trastuzumab arm as compared to the chemotherapy alone arm were neutropenia, diarrhea, fatigue, anemia, stomatitis, weight loss, upper respiratory tract infections, fever, thrombocytopenia, mucosal inflammation, nasopharyngitis, and dysgeusia. The most common adverse reactions which resulted in discontinuation of treatment on the trastuzumab-containing arm in the absence of disease progression were infection, diarrhea, and febrile neutropenia.
Adjuvant Breast Cancer
The information below reflects exposure to one-year trastuzumab therapy across three randomized, open-label studies, NSABP B31, NCCTG N9831, and HERA, with (n = 3678) or without (n = 3363) trastuzumab in the adjuvant treatment of breast cancer.
HERA
Table 3 reflects exposure to trastuzumab in 1678 patients in HERA; the median treatment duration was 51 weeks and median number of infusions was 18 [see Clinical Studies (14.1)].
Adverse Reactions | Trastuzumab (n = 1678) % | Observation (n = 1708) % |
Nervous System | ||
Headache | 10 | 3 |
Paresthesia | 2 | 0.6 |
Musculoskeletal | ||
Arthralgia | 8 | 6 |
Back pain | 5 | 3 |
Myalgia | 4 | 1 |
Bone pain | 3 | 2 |
Muscle spasm | 3 | 0.2 |
Infections | ||
Nasopharyngitis | 8 | 3 |
Urinary tract infection | 3 | 0.8 |
Gastrointestinal | ||
Diarrhea | 7 | 1 |
Nausea | 6 | 1 |
Vomiting | 3.5 | 0.6 |
Constipation | 2 | 1 |
Dyspepsia | 2 | 0.5 |
Upper abdominal pain | 2 | 1 |
General | ||
Pyrexia | 6 | 0.4 |
Peripheral edema | 5 | 2 |
Chills | 5 | 0 |
Asthenia | 4.5 | 2 |
Influenza-like illness | 2 | 0.2 |
Respiratory Thoracic Mediastinal | ||
Cough | 5 | 2 |
Influenza | 4 | 0.5 |
Dyspnea | 3 | 2 |
URI | 3 | 1 |
Rhinitis | 2 | 0.4 |
Pharyngolaryngeal pain | 2 | 0.5 |
Sinusitis | 2 | 0.3 |
Epistaxis | 2 | 0.06 |
Cardiac | ||
Hypertension | 4 | 2 |
Dizziness | 4 | 2 |
Ejection fraction decreased | 3.5 | 0.6 |
Palpitations | 3 | 0.7 |
Cardiac arrhythmias† | 3 | 1 |
Cardiac failure (congestive) | 2 | 0.3 |
Skin & Subcutaneous Tissue | ||
Rash | 4 | 0.6 |
Nail disorders | 2 | 0 |
Pruritus | 2 | 0.6 |
Clinically relevant adverse reactions in < 1% of patients who received trastuzumab in HERA included hypersensitivity (0.6%), cardiac failure (0.5%), cardiac disorder (0.3%), interstitial pneumonitis (0.2%), pulmonary hypertension (0.2%), ventricular disorder (0.2%), autoimmune thyroiditis (0.3%), and sudden death (0.06%).
Adjuvant Treatment of Breast Cancer with Trastuzumab Beyond One Year
Extending adjuvant treatment beyond one year is not recommended [see Dosage and Administration (2.3)]. In HERA, a comparison of trastuzumab administered once every 3 weeks for two years versus one year was performed. The rate of asymptomatic cardiac dysfunction was increased in the 2-year trastuzumab compared to the 1-year trastuzumab treatment arm (8.1% versus 4.6%, respectively). More patients experienced at least one adverse reaction of Grade 3 or higher in the 2-year trastuzumab treatment arm (20.4%) compared with the one-year trastuzumab treatment arm (16.3%).
NSABP B31 and NCCTG N9831
The safety data from NSABP B31 and NCCTG N9831 were obtained from 3655 patients, of whom 2000 received trastuzumab; the median treatment duration was 51 weeks [see Clinical Studies (14.1)].
In NSABP B31, only Grade 3 to 5 adverse events, treatment-related Grade 2 events, and Grade 2 to 5 dyspnea were collected during and for up to 3 months following protocol-specified treatment. The following non-cardiac adverse reactions of Grade 2 to 5 occurred at an incidence of at least 2% greater among patients receiving trastuzumab plus chemotherapy as compared to chemotherapy alone: fatigue (29.5% vs. 22.4%), infection (24.0% vs. 12.8%), hot flashes (17.1% vs. 15%), anemia (12.3% vs. 6.7%), dyspnea (11.8% vs. 4.6%), rash/desquamation (10.9% vs. 7.6%), leukopenia (10.5% vs. 8.4%), neutropenia (6.4% vs. 4.3%), headache (6.2% vs. 3.8%), pain (5.5% vs. 3%), edema (4.7% vs. 2.7%), and insomnia (4.3% vs. 1.5%). The majority of these events were Grade 2 in severity.
In NCCTG N9831, data collection was limited to the following investigator-attributed treatment-related adverse reactions: NCI-CTC Grade 4 and 5 hematologic toxicities, Grade 3 to 5 non-hematologic toxicities, selected Grade 2 to 5 toxicities associated with taxanes (myalgia, arthralgias, nail changes, motor neuropathy, and sensory neuropathy) and Grade 1 to 5 cardiac toxicities occurring during chemotherapy and/or trastuzumab treatment. The following non-cardiac adverse reactions of Grade 2 to 5 occurred at an incidence of at least 2% greater among patients receiving trastuzumab plus chemotherapy as compared to chemotherapy alone: arthralgia (12.2% vs. 9.1%), nail changes (11.5% vs. 6.8%), dyspnea (2.4% vs. 0.2%), and diarrhea (2.2% vs. 0%). The majority of these events were Grade 2 in severity.
BCIRG006
Safety data from BCIRG006 reflect exposure to trastuzumab as part of an adjuvant treatment regimen from 2124 patients receiving at least one dose of study treatment [AC-TH: n = 1068; TCH: n = 1056]. The overall median treatment duration was 54 weeks in both the AC-TH and TCH arms. The median number of infusions was 26 in the AC-TH arm and 30 in the TCH arm, including weekly infusions during the chemotherapy phase and once every three weeks dosing in the monotherapy period [see Clinical Studies (14.1)]. In BCIRG006, the toxicity profile was similar to that reported in NSABP B31, NCCTG N9831, and HERA with the exception of a lower incidence of CHF in the TCH arm.
Metastatic Breast Cancer Studies
The safety of trastuzumab was evaluated in one randomized, open-label study (H0648g) of chemotherapy with (n = 235) or without (n = 234) intravenous trastuzumab in patients with metastatic breast cancer, and in one single-arm study (H0649g) in patients with metastatic breast cancer (n = 222) [see Clinical Studies (14.1)]. Patients received 4 mg/kg initial dose of trastuzumab followed by 2 mg/kg weekly. In H0648g, 58% of patients received trastuzumab for ≥ 6 months and 9% received trastuzumab for ≥ 12 months, respectively. In H0649g, 31% of patients received trastuzumab for ≥ 6 months and 16% received trastuzumab for ≥ 12 months, respectively.
Table 4 shows the adverse reactions (≥ 5%) in patients from H0648g and H0649g.
Trastuzumab* n = 352 % | Trastuzumab + Paclitaxel % | Paclitaxel n = 95 % | Trastuzumab + AC† % | AC† % | |
General |
|
|
|
|
|
Pain | 47 | 61 | 62 | 57 | 42 |
Asthenia | 42 | 62 | 57 | 54 | 55 |
Fever | 36 | 49 | 23 | 56 | 34 |
Chills | 32 | 41 | 4 | 35 | 11 |
Headache | 26 | 36 | 28 | 44 | 31 |
Abdominal pain | 22 | 34 | 22 | 23 | 18 |
Back pain | 22 | 34 | 30 | 27 | 15 |
Infection | 20 | 47 | 27 | 47 | 31 |
Flu syndrome | 10 | 12 | 5 | 12 | 6 |
Accidental injury | 6 | 13 | 3 | 9 | 4 |
Allergic reaction | 3 | 8 | 2 | 4 | 2 |
Gastrointestinal | |||||
Nausea | 33 | 51 | 9 | 76 | 77 |
Diarrhea | 25 | 45 | 29 | 45 | 26 |
Vomiting | 23 | 37 | 28 | 53 | 49 |
Anorexia | 14 | 24 | 16 | 31 | 26 |
Nausea and vomiting | 8 | 14 | 11 | 18 | 9 |
Respiratory | |||||
Cough increased | 26 | 41 | 22 | 43 | 29 |
Dyspnea | 22 | 27 | 26 | 42 | 25 |
Rhinitis | 14 | 22 | 5 | 22 | 16 |
Pharyngitis | 12 | 22 | 14 | 30 | 18 |
Sinusitis | 9 | 21 | 7 | 13 | 6 |
Skin | |||||
Rash | 18 | 38 | 18 | 27 | 17 |
Herpes simplex | 2 | 12 | 3 | 7 | 9 |
Acne | 2 | 11 | 3 | 3 | < 1 |
Nervous | |||||
Insomnia | 14 | 25 | 13 | 29 | 15 |
Dizziness | 13 | 22 | 24 | 24 | 18 |
Paresthesia | 9 | 48 | 39 | 17 | 11 |
Depression | 6 | 12 | 13 | 20 | 12 |
Peripheral neuritis | 2 | 23 | 16 | 2 | 2 |
Neuropathy | 1 | 13 | 5 | 4 | 4 |
Metabolic | |||||
Peripheral edema | 10 | 22 | 20 | 20 | 17 |
Edema | 8 | 10 | 8 | 11 | 5 |
Cardiovascular | |||||
Congestive heart failure | 7 | 11 | 1 | 28 | 7 |
Tachycardia | 5 | 12 | 4 | 10 | 5 |
Musculoskeletal | |||||
Bone pain | 7 | 24 | 18 | 7 | 7 |
Arthralgia | 6 | 37 | 21 | 8 | 9 |
Urogenital | |||||
Urinary tract infection | 5 | 18 | 14 | 13 | 7 |
Blood and Lymphatic | |||||
Anemia | 4 | 14 | 9 | 36 | 26 |
Leukopenia | 3 | 24 | 17 | 52 | 34 |
Metastatic Gastric Cancer
The safety of trastuzumab was evaluated in patients with previously untreated for metastatic gastric or gastroesophageal junction adenocarcinoma in an open label, multi-center trial (ToGA) [see Clinical Studies (14.3)]. Patients were randomized (1:1) to receive trastuzumab in combination with cisplatin and a fluoropyrimidine (FC+H) (n=294) or chemotherapy alone (FC) (n=290). Patients in the trastuzumab plus chemotherapy arm received trastuzumab 8 mg/kg administered on Day 1 (prior to chemotherapy) followed by 6 mg/kg every 21 days until disease progression. Cisplatin was administered at 80 mg/m2 on Day 1 and the fluoropyrimidine was administered as either capecitabine 1000 mg/m2 orally twice a day on Days 1 to 14 or 5-fluorouracil 800 mg/m2/day as a continuous intravenous infusion Days 1 through 5. Chemotherapy was administered for six 21-day cycles. Median duration of trastuzumab treatment was 21 weeks and the median number of trastuzumab infusions administered was eight.
Adverse Reactions | Trastuzumab + FC | FC | ||
All Grades | Grades 3-4 | All Grades | Grades 3-4 | |
Investigations | ||||
Neutropenia | 78 | 34 | 73 | 29 |
Hypokalemia | 28 | 10 | 24 | 6 |
Anemia | 28 | 12 | 21 | 10 |
Thrombocytopenia | 16 | 5 | 11 | 3 |
Blood and Lymphatic System Disorders | ||||
Febrile Neutropenia | — | 5 | — | 3 |
Gastrointestinal Disorders | ||||
Diarrhea | 37 | 9 | 28 | 4 |
Stomatitis | 24 | 1 | 15 | 2 |
Dysphagia | 6 | 2 | 3 | < 1 |
General | ||||
Fatigue | 35 | 4 | 28 | 2 |
Fever | 18 | 1 | 12 | 0 |
Mucosal Inflammation | 13 | 2 | 6 | 1 |
Chills | 8 | < 1 | 0 | 0 |
Metabolism and Nutrition Disorders | ||||
Weight Decrease | 23 | 2 | 14 | 2 |
Infections and Infestations | ||||
Upper Respiratory Tract Infections | 19 | 0 | 10 | 0 |
Nasopharyngitis | 13 | 0 | 6 | 0 |
Renal and Urinary Disorders | ||||
Renal Failure and Impairment | 18 | 3 | 15 | 2 |
Nervous System Disorders | ||||
Dysgeusia | 10 | 0 | 5 | 0 |
The following subsections provide additional detail regarding adverse reactions observed in clinical trials of adjuvant breast cancer, metastatic breast cancer, metastatic gastric cancer, or post-marketing experience.
Cardiomyopathy
Serial measurement of cardiac function (LVEF) was obtained in clinical trials in the adjuvant treatment of breast cancer. In HERA, the median duration of follow-up was 12.6 months (12.4 months in the observation arm; 12.6 months in the 1-year trastuzumab arm); and in NSABP B31 and NCCTG N9831, 7.9 years in the AC-T arm, 8.3 years in the AC-TH arm. Following initiation of trastuzumab therapy, the incidence of new-onset dose-limiting myocardial dysfunction was higher among patients receiving trastuzumab and paclitaxel as compared to those receiving paclitaxel alone in NSABP B31 and NCCTG N9831, and in patients receiving one-year trastuzumab monotherapy compared to observation in HERA (see Table 6, Figures 1 and 2). The incidence of new-onset cardiac dysfunction, as measured by LVEF, remained similar when compared to the analysis performed at a median follow-up of 2.0 years in the AC-TH arm. This analysis showed evidence of reversibility of left ventricular dysfunction, with 64.5% of patients who experienced symptomatic CHF in the AC-TH group being asymptomatic at latest follow-up, and 90.3% having full or partial LVEF recovery.
| |||||
Study and Arm | LVEF < 50% | LVEF Decrease | |||
LVEF < 50% | ≥ 10% decrease | ≥ 16% decrease | < 20% and ≥ 10% | ≥ 20% | |
AC→TH | 23.1% | 18.5% | 11.2% | 37.9% | 8.9% |
AC→T | 11.7% | 7.0% | 3.0% | 22.1% | 3.4% |
HERA§ | |||||
Trastuzumab | 8.6% | 7.0% | 3.8% | 22.4% | 3.5% |
Observation | 2.7% | 2.0% | 1.2% | 11.9% | 1.2% |
BCIRG006¶ | |||||
TCH | 8.5% | 5.9% | 3.3% | 34.5% | 6.3% |
AC→TH | 17% | 13.3% | 9.8% | 44.3% | 13.2% |
AC→T | 9.5% | 6.6% | 3.3% | 34% | 5.5% |
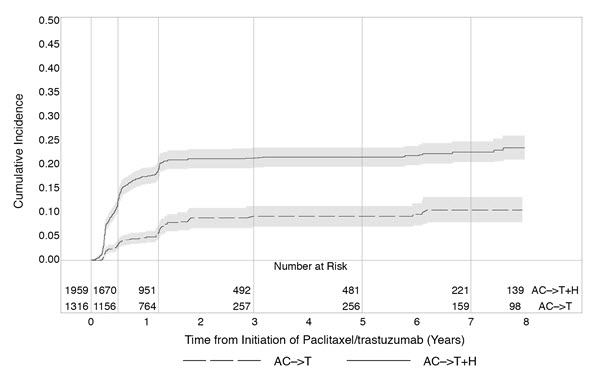
HERA: Cumulative Incidence of Time to First LVEF Decline of ≥ 10 Percentage Points from Baseline and to Below 50% with Death as a Competing Risk Event |
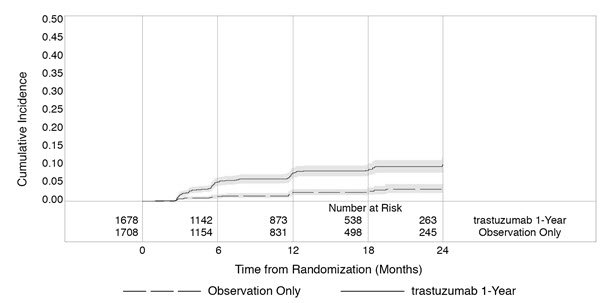 |
Time 0 is the date of randomization. |
Figure 3 BCIRG006: Cumulative Incidence of Time to First LVEF Decline of ≥ 10 Percentage Points from Baseline and to Below 50% with Death as a Competing Risk Event |
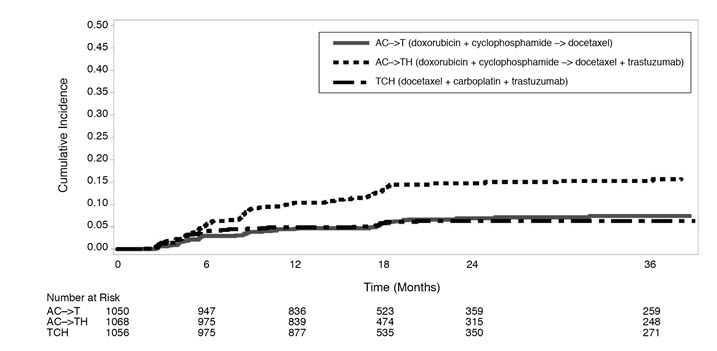 |
Time 0 is the date of randomization. |
The incidence of congestive heart failure among patients in the metastatic breast cancer trials was classified for severity using the New York Heart Association classification system (I–IV, where IV is the most severe level of cardiac failure) (see Table 2). In the metastatic breast cancer trials, the probability of cardiac dysfunction was highest in patients who received trastuzumab concurrently with anthracyclines.
In ToGA, 5% of patients in the trastuzumab plus chemotherapy arm compared to 1.1% of patients in the chemotherapy alone arm had LVEF value below 50% with a ≥ 10% absolute decrease in LVEF from pretreatment values.
Infusion Reactions
During the first infusion with trastuzumab, the symptoms most commonly reported were chills and fever, occurring in approximately 40% of patients in clinical trials. Symptoms were treated with acetaminophen, diphenhydramine, and meperidine (with or without reduction in the rate of trastuzumab infusion); permanent discontinuation of trastuzumab for infusion reactions was required in < 1% of patients. Other signs and/or symptoms may include nausea, vomiting, pain (in some cases at tumor sites), rigors, headache, dizziness, dyspnea, hypotension, elevated blood pressure, rash, and asthenia. Infusion reactions occurred in 21% and 35% of patients, and were severe in 1.4% and 9% of patients, on second or subsequent trastuzumab infusions administered as monotherapy or in combination with chemotherapy, respectively. In the post-marketing setting, severe infusion reactions, including hypersensitivity, anaphylaxis, and angioedema have been reported.
Anemia
In randomized controlled clinical trials, the overall incidence of anemia (30% vs. 21% [H0648g]), of selected NCI-CTC Grade 2 to 5 anemia (12.3% vs. 6.7% [NSABP B31]), and of anemia requiring transfusions (0.1% vs. 0 patients [NCCTG N9831]) were increased in patients receiving trastuzumab and chemotherapy compared with those receiving chemotherapy alone. Following the administration of trastuzumab as a single agent (H0649g), the incidence of NCI-CTC Grade 3 anemia was < 1%. In ToGA (metastatic gastric cancer), on the trastuzumab containing arm as compared to the chemotherapy alone arm, the overall incidence of anemia was 28% compared to 21% and of NCI-CTC Grade 3/4 anemia was 12.2% compared to 10.3%.
Neutropenia
In randomized controlled clinical trials in the adjuvant setting, the incidence of selected NCI-CTC Grade 4 to 5 neutropenia (1.7% vs. 0.8% [NCCTG N9831]) and of selected Grade 2 to 5 neutropenia (6.4% vs. 4.3% [NSABP B31]) were increased in patients receiving trastuzumab and chemotherapy compared with those receiving chemotherapy alone. In a randomized, controlled trial in patients with metastatic breast cancer, the incidences of NCI-CTC Grade 3/4 neutropenia (32% vs. 22%) and of febrile neutropenia (23% vs. 17%) were also increased in patients randomized to trastuzumab in combination with myelosuppressive chemotherapy as compared to chemotherapy alone. In ToGA (metastatic gastric cancer) on the trastuzumab containing arm as compared to the chemotherapy alone arm, the incidence of NCI-CTC Grade 3/4 neutropenia was 36.8% compared to 28.9%; febrile neutropenia 5.1% compared to 2.8%.
Infection
The overall incidences of infection (46% vs. 30% [H0648g]), of selected NCI-CTC Grade 2 to 5 infection/febrile neutropenia (24.3% vs. 13.4% [NSABP B31]) and of selected Grade 3 to 5 infection/febrile neutropenia (2.9% vs. 1.4% [NCCTG N9831]) were higher in patients receiving trastuzumab and chemotherapy compared with those receiving chemotherapy alone. The most common site of infections in the adjuvant setting involved the upper respiratory tract, skin, and urinary tract.
In BCIRG006, the overall incidence of infection was higher with the addition of trastuzumab to AC-T but not to TCH [44% (AC-TH), 37% (TCH), 38% (AC-T)]. The incidences of NCI-CTC Grade 3 to 4 infection were similar [25% (AC-TH), 21% (TCH), 23% (AC-T)] across the three arms.
In a randomized, controlled trial in treatment of metastatic breast cancer, the reported incidence of febrile neutropenia was higher (23% vs. 17%) in patients receiving trastuzumab in combination with myelosuppressive chemotherapy as compared to chemotherapy alone.
Pulmonary Toxicity
Adjuvant Breast Cancer
Among women receiving adjuvant therapy for breast cancer, the incidence of selected NCI-CTC Grade 2 to 5 pulmonary toxicity (14.3% vs. 5.4% [NSABP B31]) and of selected NCI-CTC Grade 3 to 5 pulmonary toxicity and spontaneous reported Grade 2 dyspnea (3.4% vs. 0.9% [NCCTG N9831]) was higher in patients receiving trastuzumab and chemotherapy compared with chemotherapy alone. The most common pulmonary toxicity was dyspnea (NCI-CTC Grade 2 to 5: 11.8% vs. 4.6% [NSABP B31]; NCI-CTC Grade 2 to 5: 2.4% vs. 0.2% [NCCTG N9831]).
Pneumonitis/pulmonary infiltrates occurred in 0.7% of patients receiving trastuzumab compared with 0.3% of those receiving chemotherapy alone. Fatal respiratory failure occurred in 3 patients receiving trastuzumab, one as a component of multi-organ system failure, as compared to 1 patient receiving chemotherapy alone.
In HERA, there were 4 cases of interstitial pneumonitis in the one-year trastuzumab treatment arm compared to none in the observation arm at a median follow-up duration of 12.6 months.
Metastatic Breast Cancer
Among women receiving trastuzumab for treatment of metastatic breast cancer, the incidence of pulmonary toxicity was also increased. Pulmonary adverse events have been reported in the post-marketing experience as part of the symptom complex of infusion reactions. Pulmonary events include bronchospasm, hypoxia, dyspnea, pulmonary infiltrates, pleural effusions, non-cardiogenic pulmonary edema, and acute respiratory distress syndrome. For a detailed description, see Warnings and Precautions (5.4).
Thrombosis/Embolism
In 4 randomized, controlled clinical trials, the incidence of thrombotic adverse events was higher in patients receiving trastuzumab and chemotherapy compared to chemotherapy alone in three studies (2.6% vs. 1.5% [NSABP B31], 2.5% and 3.7% vs. 2.2% [BCIRG006] and 2.1% vs. 0% [H0648g]).
Diarrhea
Among women receiving adjuvant therapy for breast cancer, the incidence of NCI-CTC Grade 2 to 5 diarrhea (6.7% vs. 5.4% [NSABP B31]) and of NCI-CTC Grade 3 to 5 diarrhea (2.2% vs. 0% [NCCTG N9831]), and of Grade 1 to 4 diarrhea (7% vs. 1% [HERA; one-year trastuzumab treatment at 12.6 months median duration of follow-up]) were higher in patients receiving trastuzumab as compared to controls. In BCIRG006, the incidence of Grade 3 to 4 diarrhea was higher [5.7% AC-TH, 5.5% TCH vs. 3.0% AC-T] and of Grade 1 to 4 was higher [51% AC-TH, 63% TCH vs. 43% AC-T] among women receiving trastuzumab. Of patients receiving trastuzumab as a single agent for the treatment of metastatic breast cancer, 25% experienced diarrhea. An increased incidence of diarrhea was observed in patients receiving trastuzumab in combination with chemotherapy for treatment of metastatic breast cancer.
Renal Toxicity
In ToGA (metastatic gastric cancer) on the trastuzumab-containing arm as compared to the chemotherapy alone arm the incidence of renal impairment was 18% compared to 14.5%. Severe (Grade 3/4) renal failure was 2.7% on the trastuzumab-containing arm compared to 1.7% on the chemotherapy only arm. Treatment discontinuation for renal insufficiency/failure was 2% on the trastuzumab-containing arm and 0.3% on the chemotherapy only arm.
In the post-marketing setting, rare cases of nephrotic syndrome with pathologic evidence of glomerulopathy have been reported. The time to onset ranged from 4 months to approximately 18 months from initiation of trastuzumab therapy. Pathologic findings included membranous glomerulonephritis, focal glomerulosclerosis, and fibrillary glomerulonephritis. Complications included volume overload and congestive heart failure.
6.2 Post-Marketing Experience
The following adverse reactions have been identified during post-approval use of trastuzumab products. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- •
- Infusion reaction [see Warnings and Precautions (5.2)]
- •
- Oligohydramnios or oligohydramnios sequence, including pulmonary hypoplasia, skeletal abnormalities, and neonatal death [see Warnings and Precautions (5.3)]
- •
- Glomerulopathy [see Adverse Reactions (6.1)]
- •
- Immune thrombocytopenia
- •
- Tumor lysis syndrome (TLS): Cases of possible TLS have been reported in patients treated with trastuzumab products. Patients with significant tumor burden (e.g. bulky metastases) may be at a higher risk. Patients could present with hyperuricemia, hyperphosphatemia, and acute renal failure which may represent possible TLS. Providers should consider additional monitoring and/or treatment as clinically indicated.
Additional Resources
Chat online with Pfizer Medical Information regarding your inquiry on a Pfizer medicine or vaccine.
Speak with a Pfizer Medical Information Professional regarding your Pfizer medicine or vaccine inquiry.
Available 9AM-5PM ET Monday to Friday; excluding holidays.
Submit a medical question for a Pfizer medicine or a vaccine.
The submission will be reviewed during our standard business hours.
To report an adverse event related to a Pfizer product and you are not part of a clinical trial* for this medication, click the link below to submit your information:
Pfizer Safety Reporting Site
*If you are involved in a clinical trial for either product, adverse events should be reported to your coordinating study site.
If you cannot use the above website to report an adverse event related to a Pfizer medication, please call (800) 438-1985.
You may also contact the U.S. Food and Drug Administration (FDA) directly to report adverse events or product quality concerns either online at www.fda.gov/medwatch or by calling (800) 332-1088.