(nirmatrelvir tablets; ritonavir tablets)
8 USE IN SPECIFIC POPULATIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data on the use of nirmatrelvir during pregnancy are insufficient to evaluate for a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. Published observational studies on ritonavir use in pregnant women have not identified an increase in the risk of major birth defects. Published studies with ritonavir are insufficient to identify a drug associated risk of miscarriage (see Data). There are maternal and fetal risks associated with untreated COVID-19 in pregnancy (see Clinical Considerations).
In an embryo-fetal development study with nirmatrelvir, reduced fetal body weights following oral administration of nirmatrelvir to pregnant rabbits were observed at systemic exposures (AUC) approximately 11 times higher than clinical exposure at the approved human dose of PAXLOVID. No other adverse developmental outcomes were observed in animal reproduction studies with nirmatrelvir at systemic exposures (AUC) greater than or equal to 3 times higher than clinical exposure at the approved human dose of PAXLOVID (see Data).
In embryo-fetal developmental studies with ritonavir, no evidence of adverse developmental outcomes was observed following oral administration of ritonavir to pregnant rats and rabbits at systemic exposures (AUC) 5 (rat) or 8 (rabbits) times higher than clinical exposure at the approved human dose of PAXLOVID (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Human Data
Ritonavir
Based on prospective reports to the antiretroviral pregnancy registry of live births following exposure to ritonavir-containing regimens (including over 3,500 live births exposed in the first-trimester and over 3,500 live births exposed in the second and third trimesters), there was no difference in the rate of overall birth defects for ritonavir compared with the background birth defect rate of 2.7% in the U.S. reference population of the Metropolitan Atlanta Congenital Defects Program (MACDP). The prevalence of birth defects in live births was 2.4% [95% confidence interval (CI): 1.9%, 2.9%] following first-trimester exposure to ritonavir-containing regimens and 2.9% (95% CI: 2.4%, 3.5%) following second and third trimester exposure to ritonavir-containing regimens. While placental transfer of ritonavir and fetal ritonavir concentrations are generally low, detectable levels have been observed in cord blood samples and neonate hair.
Animal Data
Nirmatrelvir
Embryo-fetal developmental (EFD) toxicity studies were conducted in pregnant rats and rabbits administered oral nirmatrelvir doses of up to 1,000 mg/kg/day during organogenesis [on Gestation Days (GD) 6 through 17 in rats and GD 7 through 19 in rabbits]. No biologically significant developmental effects were observed in the rat EFD study. At the highest dose of 1,000 mg/kg/day, the systemic nirmatrelvir exposure (AUC24) in rats was approximately 9 times higher than clinical exposures at the approved human dose of PAXLOVID. In the rabbit EFD study, lower fetal body weights (9% decrease) were observed at 1,000 mg/kg/day in the absence of significant maternal toxicity findings. At 1,000 mg/kg/day, the systemic exposure (AUC24) in rabbits was approximately 11 times higher than clinical exposures at the approved human dose of PAXLOVID. No other significant developmental toxicities (malformations and embryo-fetal lethality) were observed up to the highest dose tested, 1,000 mg/kg/day. No developmental effects were observed in rabbits at 300 mg/kg/day resulting in systemic exposure (AUC24) approximately 3 times higher than clinical exposures at the approved human dose of PAXLOVID. A pre- and postnatal developmental (PPND) study in pregnant rats administered oral nirmatrelvir doses of up to 1,000 mg/kg/day from GD 6 through Lactation Day (LD) 20 showed no adverse findings. Although no difference in body weight was noted at birth when comparing offspring born to nirmatrelvir-treated versus control animals, a decrease in the body weight of offspring was observed on Postnatal Day (PND) 17 (8% decrease) and PND 21 (up to 7% decrease) in the absence of maternal toxicity. No significant differences in offspring body weight were observed from PND 28 to PND 56. The maternal systemic exposure (AUC24) at 1,000 mg/kg/day was approximately 9 times higher than clinical exposures at the approved human dose of PAXLOVID. No body weight changes in the offspring were noted at 300 mg/kg/day, where maternal systemic exposure (AUC24) was approximately 6 times higher than clinical exposures at the approved human dose of PAXLOVID.
Ritonavir
Ritonavir was administered orally to pregnant rats (at 0, 15, 35, and 75 mg/kg/day) and rabbits (at 0, 25, 50, and 110 mg/kg/day) during organogenesis (on GD 6 through 17 in rats and GD 6 through 19 in rabbits). No evidence of teratogenicity due to ritonavir was observed in rats and rabbits at systemic exposures (AUC) 5 (rats) or 8 (rabbits) times higher than exposure at the approved human dose of PAXLOVID. Increased incidences of early resorptions, ossification delays, and developmental variations, as well as decreased fetal body weights were observed in rats in the presence of maternal toxicity, at systemic exposures (AUC) approximately 10 times higher than exposure at the approved human dose of PAXLOVID. In rabbits, resorptions, decreased litter size, and decreased fetal weights were observed at maternally toxic doses, at systemic exposures greater than 8 times higher than exposure at the approved human dose of PAXLOVID. In a PPND study in rats, administration of 0, 15, 35, and 60 mg/kg/day ritonavir from GD 6 through PND 20 resulted in no developmental toxicity, at ritonavir systemic exposures greater than 10 times the exposure at the approved human dose of PAXLOVID.
8.2 Lactation
Risk Summary
Nirmatrelvir and ritonavir are present in human breast milk in small amounts (less than 2%). In a clinical lactation study in 8 lactating women, nirmatrelvir and ritonavir were estimated to be present in human milk at a mean weight-normalized infant daily dose of 0.16 mg/kg/day (1.8% of maternal weight-adjusted daily dose) and 0.006 mg/kg/day (0.2% of maternal weight-adjusted daily dose), respectively (see Data).
There are no available data on the effects of nirmatrelvir or ritonavir on the breastfed infant or on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for PAXLOVID and any potential adverse effects on the breastfed infant from PAXLOVID or from the underlying maternal condition. Breastfeeding individuals with COVID-19 should follow practices according to clinical guidelines to avoid exposing the infant to COVID-19.
In a clinical pharmacokinetics study, 8 healthy lactating women who were at least 12 weeks postpartum were administered 3 oral doses every 12 hours (steady state dosing) of 300 mg/100 mg nirmatrelvir/ritonavir. The mean daily amount of nirmatrelvir and ritonavir recovered in breast milk was 0.752 mg and 0.027 mg, respectively, representing 0.13% and 0.014% of the corresponding administered daily maternal doses (unadjusted for weight). The estimated daily infant dose (assuming average milk consumption of 150 mL/kg/day), was 0.16 mg/kg/day and 0.006 mg/kg/day, 1.8% and 0.2% of the maternal dose, respectively, for nirmatrelvir and ritonavir.
8.3 Females and Males of Reproductive Potential
Contraception
Use of ritonavir may reduce the efficacy of combined hormonal contraceptives. Advise patients using combined hormonal contraceptives to use an effective alternative contraceptive method or an additional barrier method of contraception [see Drug Interactions (7.3)].
8.5 Geriatric Use
Clinical studies of PAXLOVID include subjects 65 years of age and older and their data contributes to the overall assessment of safety and efficacy [see Adverse Reactions (6.1) and Clinical Studies (14.1)]. Of the total number of subjects in the integrated dataset consisting of EPIC-HR and EPIC-SR who were randomized to and received PAXLOVID (N=1,578), 165 (10%) were 65 years of age and older and 39 (2%) were 75 years of age and older. No overall differences in safety were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in safety between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
8.6 Renal Impairment
Renal impairment increases nirmatrelvir exposure, which may increase the risk of PAXLOVID adverse reactions. No dosage adjustment is recommended in patients with mild renal impairment (eGFR ≥60 to <90 mL/min). Reduce the PAXLOVID dosage in patients with moderate renal impairment (eGFR ≥30 to <60 mL/min). Reduce the PAXLOVID dose and dose frequency in patients with severe renal impairment (eGFR <30 mL/min), including those requiring hemodialysis. On days when patients undergo hemodialysis, the PAXLOVID dose should be administered after hemodialysis [see Dosage and Administration (2.3), Adverse Reactions (6.1), and Clinical Pharmacology (12.3)]. Prescriptions should specify the numeric dose of each active ingredient within PAXLOVID. Providers should counsel patients about renal dosing instructions [see Patient Counseling Information (17)].
8.7 Hepatic Impairment
No dosage adjustment of PAXLOVID is recommended for patients with mild (Child-Pugh Class A) or moderate (Child-Pugh Class B) hepatic impairment. No pharmacokinetic or safety data are available regarding the use of nirmatrelvir or ritonavir in subjects with severe (Child-Pugh Class C) hepatic impairment, therefore, PAXLOVID is not recommended for use in patients with severe (Child-Pugh Class C) hepatic impairment [see Warnings and Precautions (5.3) and Clinical Pharmacology (12.3)].
MEDICATION GUIDE
PATIENT INFORMATION | ||||||
What is the most important information I should know about PAXLOVID? PAXLOVID can interact with other medicines causing severe or life-threatening side effects or death. It is important to know the medicines that should not be taken with PAXLOVID. Do not take PAXLOVID if:
| ||||||
|
|
| ||||
These are not the only medicines that may cause serious or life-threatening side effects if taken with PAXLOVID. PAXLOVID may increase or decrease the levels of multiple other medicines. It is very important to tell your healthcare provider about all of the medicines you are taking because additional laboratory tests or changes in the dose of your other medicines may be necessary during treatment with PAXLOVID. Your healthcare provider may also tell you about specific symptoms to watch out for that may indicate that you need to stop or decrease the dose of some of your other medicines.
| ||||||
What is PAXLOVID? PAXLOVID is not approved for use as pre-exposure or post-exposure treatment for prevention of COVID-19. | ||||||
Before taking PAXLOVID, tell your healthcare provider about all of your medical conditions, including if you:
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
Tell your healthcare provider if you are taking combined birth control (hormonal contraceptive). PAXLOVID may affect how your hormonal contraceptives work. Females who are able to become pregnant should use another effective alternative form of contraception or an additional barrier method of contraception during treatment with PAXLOVID. Talk to your healthcare provider if you have any questions about contraceptive methods that might be right for you. | ||||||
How should I take PAXLOVID?
| ||||||
If you are prescribed PAXLOVID 300 mg; 100 mg Dose Pack | ||||||
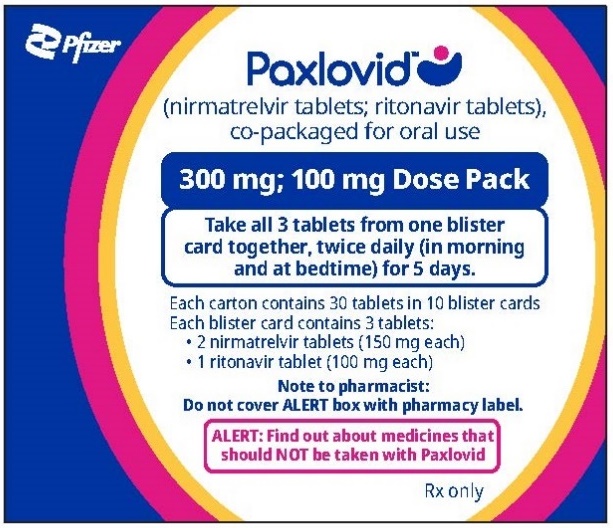 | ||||||
How to take PAXLOVID 300 mg; 100 mg Dose Pack | ||||||
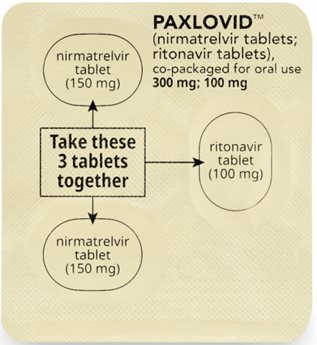 | Morning Dose: | |||||
 | ||||||
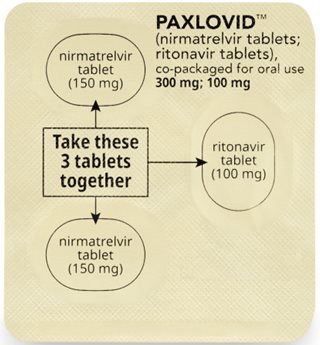 | Bedtime Dose: | |||||
 | ||||||
If you are prescribed PAXLOVID 150 mg; 100 mg Dose Pack | ||||||
 | ||||||
How to take PAXLOVID 150 mg; 100 mg Dose Pack | ||||||
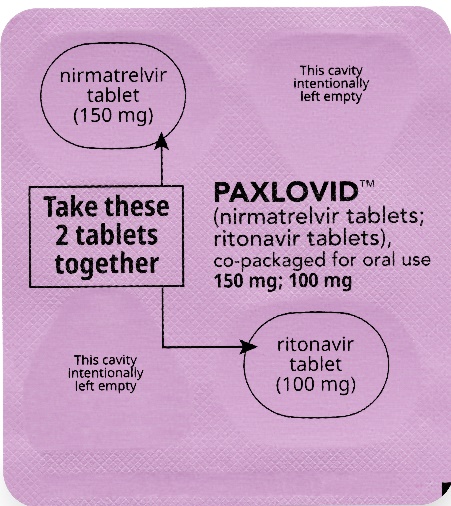 | Morning Dose: | |||||
 | ||||||
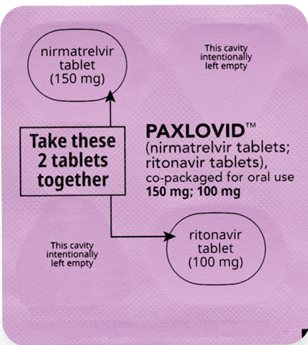 | Bedtime Dose: | |||||
 | ||||||
If you are prescribed PAXLOVID 300 mg; 100 mg (Day 1) and 150 mg; 100 mg (Days 2-5) | ||||||
 | ||||||
How to take PAXLOVID 300 mg; 100 mg (Day 1) and 150 mg; 100 mg (Days 2-5) | ||||||
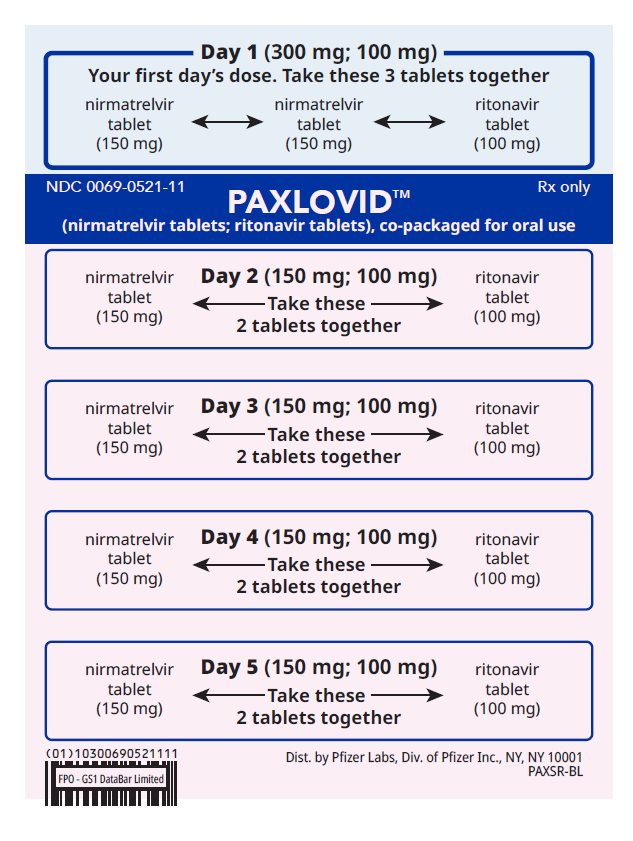 | Day 1 (First Day): Take the 2 pink nirmatrelvir tablets and | |||||
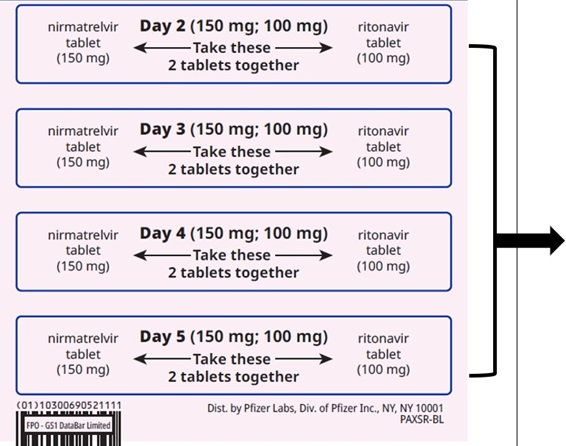 | Days 2-5: Take the 1 pink nirmatrelvir tablet and 1 white ritonavir tablet together | |||||
Talk to your healthcare provider if you do not feel better or if you feel worse after 5 days. | ||||||
What are the possible side effects of PAXLOVID? PAXLOVID may cause serious side effects, including:
| ||||||
|
| |||||
| ||||||
|
| |||||
The most common side effects of PAXLOVID include: altered sense of taste (such as metallic, bitter taste) and diarrhea. Other possible side effects include:
These are not all of the possible side effects of PAXLOVID. For more information, ask your healthcare provider or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. | ||||||
How should I store PAXLOVID?
Keep PAXLOVID and all medicines out of the reach of children. | ||||||
General information about the safe and effective use of PAXLOVID. | ||||||
What are the ingredients in PAXLOVID? | ||||||
 LAB-1524-5.0 | ||||||


This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 02/2026
Additional Resources
Chat online with Pfizer Medical Information regarding your inquiry on a Pfizer medicine or vaccine.
Speak with a Pfizer Medical Information Professional regarding your Pfizer medicine or vaccine inquiry.
Available 9AM-5PM ET Monday to Friday; excluding holidays.
Submit a medical question for a Pfizer medicine or a vaccine.
The submission will be reviewed during our standard business hours.
To report an adverse event related to a Pfizer product and you are not part of a clinical trial* for this medication, click the link below to submit your information:
Pfizer Safety Reporting Site
*If you are involved in a clinical trial for either product, adverse events should be reported to your coordinating study site.
If you cannot use the above website to report an adverse event related to a Pfizer medication, please call (800) 438-1985.
You may also contact the U.S. Food and Drug Administration (FDA) directly to report adverse events or product quality concerns either online at www.fda.gov/medwatch or by calling (800) 332-1088.