(voxelotor)
14 CLINICAL STUDIES
14 CLINICAL STUDIES
14.1 Adults and Pediatric Patients 12 Years and Older
The efficacy and safety of OXBRYTA in SCD was evaluated in HOPE, a Phase 3 randomized, double-blind, placebo-controlled, multicenter trial [NCT 03036813]. In this study, 274 patients were randomized to daily oral administration of OXBRYTA 1,500 mg (N=90), OXBRYTA 900 mg (N=92), or placebo (N=92). Patients were included if they had from 1 to 10 vasoocclusive crisis (VOC) events within 12 months prior to enrollment and baseline hemoglobin (Hb) ≥5.5 to ≤10.5 g/dL. Eligible patients on stable doses of hydroxyurea for at least 90 days were allowed to continue hydroxyurea therapy throughout the study. Randomization was stratified by patients already receiving hydroxyurea (yes, no), geographic region (North America, Europe, Other), and age (12 to <17 years, 18 to 65 years). The trial excluded patients who received red blood cell (RBC) transfusions within 60 days and erythropoietin within 28 days of enrollment, had renal insufficiency, uncontrolled liver disease, were pregnant, or lactating.
The majority of patients had HbSS or HbS/beta0-thalassemia genotype (90%) and were receiving background hydroxyurea therapy (65%). The median age was 24 years (range: 12 to 64 years); 46 (17%) patients were 12 to <17 years. Median baseline Hb was 8.5 g/dL (5.9 to 10.8 g/dL). One hundred and fifteen (42%) had 1 VOC event and 159 (58%) had 2 to 10 events within 12 months prior to enrollment. In the OXBRYTA 1,500 mg group, 63 (70%) patients completed the study through Week 72.
Efficacy was based on Hb response rate defined as a Hb increase of >1 g/dL from baseline to Week 24 in patients treated with OXBRYTA 1,500 mg versus placebo. The response rate for OXBRYTA 1,500 mg was 51.1% (46/90) compared to 6.5% (6/92) in the placebo group (p < 0.001). No outlier subgroups were observed. The distribution of Hb change from baseline for individual patients completing 24 weeks of treatment with OXBRYTA 1,500 mg or placebo is depicted in Figure 1.
|
Figure 1: Subject-level Change from Baseline in Hemoglobin at Week 24 in Patients Who Completed 24 Weeks of Treatment* |
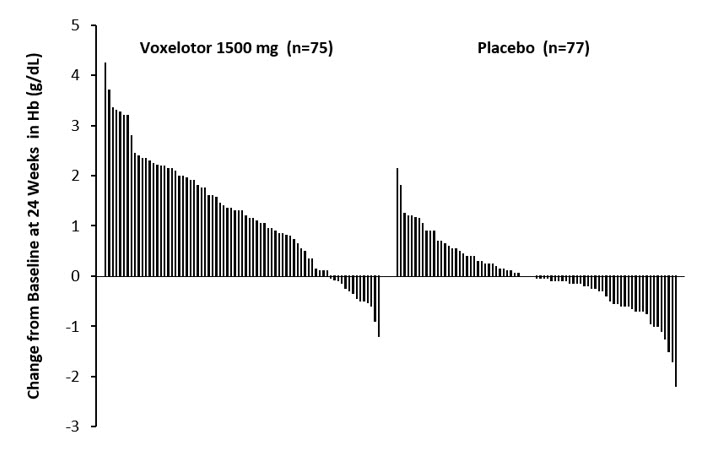 |
Additional efficacy evaluation included change in Hb and percent change in indirect bilirubin and percent reticulocyte count from baseline to Week 24 (Table 6).
| OXBRYTA 1,500 mg QD (N=90) | Placebo (N=92) | P Value | |
|---|---|---|---|
| QD = once daily; SE = standard error | |||
Hemoglobin | 1.1 g/dL | -0.1 g/dL | < 0.001 |
Indirect Bilirubin | -29.1% | -2.8% | < 0.001 |
Percent Reticulocyte Count | -18.0% | 6.8% | < 0.001 |
14.2 Pediatric Patients 4 to <12 Years
The efficacy and safety of OXBRYTA in patients 4 to <12 years with SCD was evaluated in an open-label, multi-center, Phase 2 trial [NCT 02850406]. In this study, 45 patients 4 to <12 years and 11 patients 12 to <17 years received OXBRYTA. Patients 4 to <12 years received tablets for oral suspension based on body weight at baseline. OXBRYTA doses of 600 mg, 900 mg, or 1,500 mg once daily were administered to patients weighing 10 kg to <20 kg, 20 kg to <40 kg, or ≥40 kg, respectively. Patients 12 to <17 years received OXBRYTA 1,500 mg once daily.
Patients were included if their baseline hemoglobin (Hb) was ≤10.5 g/dL. Eligible patients on stable doses of hydroxyurea for at least 90 days were allowed to continue hydroxyurea therapy throughout the study. The trial excluded patients who had a VOC event within 14 days prior to enrollment, received red blood cell (RBC) transfusions within 30 days of enrollment, and had renal insufficiency or uncontrolled liver disease.
All patients had HbSS or HbS/beta0-thalassemia genotype (100%) and most were receiving background hydroxyurea therapy (80%). The median age was 8 years (range: 4 to 15 years); 45 (80%) patients were 4 to <12 years. In this age group, mean baseline Hb was 8.6 g/dL (range: 6.1 to 10.5 g/dL).
Efficacy was based on Hb response rate, which is defined as a Hb increase of >1 g/dL from baseline to Week 24. Hb response rate for OXBRYTA in patients aged 4 to <12 years who took at least one dose of OXBRYTA was 36% (16/45) (95% CI: 21.6%, 49.5%).
MEDICATION GUIDE
This Patient Information has been approved by the U.S. Food and Drug Administration | Revised: 08/2023 | |||||
PATIENT INFORMATION | ||||||
OXBRYTA® (ox brye ta) |
| OXBRYTA® (ox brye ta) | ||||
What is OXBRYTA? | ||||||
Do not take OXBRYTA if you or your child have had an allergic reaction to voxelotor or any of the ingredients in OXBRYTA. See the end of this leaflet for a list of the ingredients in OXBRYTA. | ||||||
Before taking OXBRYTA, tell your healthcare provider about all of your medical conditions, including if you or your child:
Tell your healthcare provider about all the medicines you or your child take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Some medicines may affect how OXBRYTA works. OXBRYTA may also affect how other medicines work and may affect the results of certain blood tests. Keep a list of all your medicines and show it to your healthcare provider. | ||||||
How should I take OXBRYTA?
| ||||||
What should I avoid while taking OXBRYTA? | ||||||
What are the possible side effects of OXBRYTA?
| ||||||
|
| |||||
The most common side effects of OXBRYTA include: | ||||||
|
| |||||
The most common side effects of OXBRYTA in children ages 4 to less than 12 years of age include: | ||||||
|
| |||||
These are not all the possible side effects of OXBRYTA. | ||||||
How should I store OXBRYTA?
Keep OXBRYTA and all medicines out of the reach of children. | ||||||
General information about the safe and effective use of OXBRYTA. | ||||||
What are the ingredients of OXBRYTA? Distributed by Global Blood Therapeutics, Inc A subsidiary of Pfizer Inc. South San Francisco, CA 94080 For more information, go to www.pfizer.com or call 1-800-438-1985. | ||||||

Additional Resources
Chat online with Pfizer Medical Information regarding your inquiry on a Pfizer medicine or vaccine.
Speak with a Pfizer Medical Information Professional regarding your Pfizer medicine or vaccine inquiry.
Available 9AM-5PM ET Monday to Friday; excluding holidays.
Submit a medical question for a Pfizer medicine or a vaccine.
The submission will be reviewed during our standard business hours.
To report an adverse event related to a Pfizer product and you are not part of a clinical trial* for this medication, click the link below to submit your information:
Pfizer Safety Reporting Site
*If you are involved in a clinical trial for either product, adverse events should be reported to your coordinating study site.
If you cannot use the above website to report an adverse event related to a Pfizer medication, please call (800) 438-1985.
You may also contact the U.S. Food and Drug Administration (FDA) directly to report adverse events or product quality concerns either online at www.fda.gov/medwatch or by calling (800) 332-1088.