(infliximab-dyyb)
14 CLINICAL STUDIES
14 CLINICAL STUDIES
14.1 Adult Crohn's Disease
Active Crohn's Disease in Adults
The safety and efficacy of single and multiple doses of infliximab were assessed in 2 randomized, double-blind, placebo-controlled clinical studies in 653 adult patients with moderate to severely active CD [Crohn's Disease Activity Index (CDAI) ≥220 and ≤400] with an inadequate response to prior conventional therapies. Concomitant stable doses of aminosalicylates, corticosteroids and/or immunomodulatory agents were permitted and 92% of patients continued to receive at least one of these medications.
In the single-dose trial of 108 adult patients, 16% (4/25) of placebo patients achieved a clinical response (decrease in CDAI ≥70 points) at Week 4 vs. 81% (22/27) of patients receiving 5 mg/kg infliximab (P<0.001, two-sided, Fisher's Exact test). Additionally, 4% (1/25) of placebo patients and 48% (13/27) of patients receiving 5 mg/kg of infliximab achieved clinical remission (CDAI<150) at Week 4.
In a multidose trial (ACCENT I [Study Crohn's I]), 545 adult patients received 5 mg/kg at Week 0 and were then randomized to one of three treatment groups; the placebo maintenance group received placebo at Weeks 2 and 6, and then every 8 weeks; the 5 mg/kg maintenance group received 5 mg/kg at Weeks 2 and 6, and then every 8 weeks; and the 10 mg/kg maintenance group received 5 mg/kg at Weeks 2 and 6, and then 10 mg/kg every 8 weeks. Patients in response at Week 2 were randomized and analyzed separately from those not in responses at Week 2. Corticosteroid taper was permitted after Week 6.
At Week 2, 57% (311/545) of patients were in clinical response. At Week 30, a significantly greater proportion of these patients in the 5 mg/kg and 10 mg/kg maintenance groups achieved clinical remission compared to patients in the placebo maintenance group (Table 3).
Additionally, a significantly greater proportion of patients in the 5 mg/kg and 10 mg/kg infliximab maintenance groups were in clinical remission and were able to discontinue corticosteroid use compared to patients in the placebo maintenance group at Week 54 (Table 3).
| Single 5-mg/kg Dose* | Three-Dose Induction† | ||
|---|---|---|---|
| Placebo Maintenance | Infliximab Maintenance q 8wks | ||
| 5 mg/kg | 10 mg/kg | ||
Week 30 | |||
Clinical remission | 25/102 | 41/104 | 48/105 |
P-value‡ | 0.022 | 0.001 | |
Week 54 | |||
Patients in remission able to discontinue corticosteroid use§ | 6/54 | 14/56 | 18/53 |
P-value‡ | 0.059 | 0.005 | |
Patients in the infliximab maintenance groups (5 mg/kg and 10 mg/kg) had a longer time to loss of response than patients in the placebo maintenance group (Figure 1). At Weeks 30 and 54, significant improvement from baseline was seen among the 5 mg/kg and 10 mg/kg groups treated with infliximab compared to the placebo group in the disease-specific inflammatory bowel disease questionnaire (IBDQ), particularly the bowel and systemic components, and in the physical component summary score of the general health-related quality of life questionnaire SF-36.
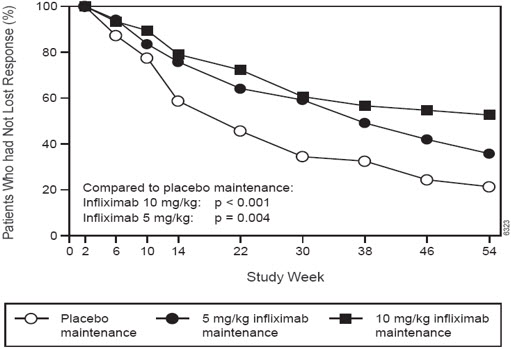
Figure 1 Kaplan-Meier Estimate of the Proportion of Adults with CD Who Had Not Lost Response Through Week 54 (Study Crohn's I)
In a subset of 78 patients who had mucosal ulceration at baseline and who participated in an endoscopic substudy, 13 of 43 patients in infliximab maintenance group had endoscopic evidence of mucosal healing compared to 1 of 28 patients in the placebo group at Week 10. Of the patients treated with infliximab showing mucosal healing at Week 10, 9 of 12 patients also showed mucosal healing at Week 54.
Patients who achieved a response and subsequently lost response were eligible to receive infliximab on an episodic basis at a dose that was 5 mg/kg higher than the dose to which they were randomized. The majority of such patients responded to the higher dose. Among patients who were not in response at Week 2, 59% (92/157) of maintenance patients on infliximab responded by Week 14 compared to 51% (39/77) of placebo maintenance patients. Among patients who did not respond by Week 14, additional therapy did not result in significantly more responses [see Dosage and Administration (2)].
Fistulizing Crohn's Disease in Adults
The safety and efficacy of infliximab were assessed in 2 randomized, double-blind, placebo-controlled studies in adult patients with fistulizing CD with fistula(s) that were of at least 3 months duration. Concurrent use of stable doses of corticosteroids, 5-aminosalicylates, antibiotics, MTX, 6-MP and/or AZA was permitted.
In the first trial, 94 adult patients received 3 doses of either placebo or infliximab at Weeks 0, 2 and 6. Fistula response (≥50% reduction in number of enterocutaneous fistulas draining upon gentle compression on at least 2 consecutive visits without an increase in medication or surgery for CD) was seen in 68% (21/31) of patients in the 5 mg/kg infliximab group (P=0.002) and 56% (18/32) of patients in the 10 mg/kg infliximab group (P=0.021) vs. 26% (8/31) of patients in the placebo arm. The median time to onset of response and median duration of response in patients treated with infliximab was 2 and 12 weeks, respectively. Closure of all fistulas was achieved in 52% patients treated with infliximab compared with 13% of placebo-treated patients (P<0.001).
In the second trial (ACCENT II [Study Crohn's II]), adult patients who were enrolled had to have at least 1 draining enterocutaneous (perianal, abdominal) fistula. All patients received 5 mg/kg of infliximab at Weeks 0, 2 and 6. Patients were randomized to placebo or 5 mg/kg maintenance with infliximab at Week 14. Patients received maintenance doses at Week 14 and then every 8 weeks through Week 46. Patients who were in fistula response (fistula response was defined the same as in the first trial) at both Weeks 10 and 14 were randomized separately from those not in response. The primary endpoint was time from randomization to loss of response among those patients who were in fistula response.
Among the randomized patients (273 of the 296 initially enrolled), 87% had perianal fistulas and 14% had abdominal fistulas. Eight percent also had rectovaginal fistulas. Greater than 90% of the patients had received previous immunosuppressive and antibiotic therapy.
At Week 14, 65% (177/273) of patients were in fistula response. Patients randomized to maintenance with infliximab had a longer time to loss of fistula response compared to the placebo maintenance group (Figure 2). At Week 54, 38% (33/87) of patients treated with infliximab had no draining fistulas compared with 22% (20/90) of placebo-treated patients (P=0.02). Compared to placebo maintenance, patients on maintenance treatment with infliximab had a trend toward fewer hospitalizations.
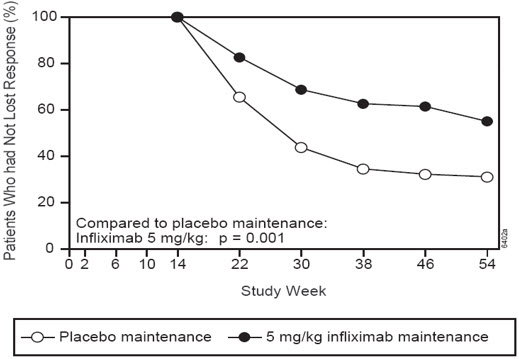
Figure 2 Life Table Estimates of the Proportion of Adult CD Patients Who Had Not Lost Fistula Response Through Week 54 (Study Crohn's II)
Patients who achieved a fistula response and subsequently lost response were eligible to receive maintenance therapy with infliximab at a dose that was 5 mg/kg higher than the dose to which they were randomized. Of the placebo maintenance patients, 66% (25/38) responded to 5 mg/kg infliximab, and 57% (12/21) of maintenance patients on infliximab responded to 10 mg/kg.
Patients who had not achieved a response by Week 14 were unlikely to respond to additional doses of infliximab.
Similar proportions of patients in either group developed new fistulas (17% overall) and similar numbers developed abscesses (15% overall).
14.2 Pediatric Crohn's Disease
The safety and efficacy of infliximab were assessed in a randomized, open-label study (Study Peds Crohn's) in 112 pediatric patients aged 6 to 17 years old with moderately to severely active CD and an inadequate response to conventional therapies. The median age was 13 years and the median Pediatric Crohn's Disease Activity Index (PCDAI) was 40 (on a scale of 0 to 100). All patients were required to be on a stable dose of 6-MP, AZA, or MTX; 35% were also receiving corticosteroids at baseline.
All patients received induction dosing of 5 mg/kg of infliximab at Weeks 0, 2, and 6. At Week 10, 103 patients were randomized to a maintenance regimen of 5 mg/kg of infliximab given either every 8 weeks or every 12 weeks.
At Week 10, 88% of patients were in clinical response (defined as a decrease from baseline in the PCDAI score of ≥15 points and total PCDAI score of ≤30 points), and 59% were in clinical remission (defined as PCDAI score of ≤10 points).
The proportion of pediatric patients achieving clinical response at Week 10 compared favorably with the proportion of adults achieving a clinical response in Study Crohn's I. The study definition of clinical response in Study Peds Crohn's was based on the PCDAI score, whereas the CDAI score was used in the adult Study Crohn's I.
At both Week 30 and Week 54, the proportion of patients in clinical response was greater in the every 8-week treatment group than in the every 12-week treatment group (73% vs. 47% at Week 30, and 64% vs. 33% at Week 54). At both Week 30 and Week 54, the proportion of patients in clinical remission was also greater in the every 8-week treatment group than in the every 12-week treatment group (60% vs. 35% at Week 30, and 56% vs. 24% at Week 54), (Table 4).
For patients in Study Peds Crohn's receiving corticosteroids at baseline, the proportion of patients able to discontinue corticosteroids while in remission at Week 30 was 46% for the every 8-week maintenance group and 33% for the every 12-week maintenance group. At Week 54, the proportion of patients able to discontinue corticosteroids while in remission was 46% for the every 8-week maintenance group and 17% for the every 12-week maintenance group.
| 5 mg/kg Infliximab | ||
|---|---|---|
| Every 8 Week Treatment Group | Every 12 Week Treatment Group | |
Patients randomized | 52 | 51 |
Clinical Response* | ||
Week 30 | 73%† | 47% |
Week 54 | 64%† | 33% |
Clinical Remission‡ | ||
Week 30 | 60%§ | 35% |
Week 54 | 56%† | 24% |
14.3 Adult Ulcerative Colitis
The safety and efficacy of infliximab were assessed in 2 randomized, double-blind, placebo-controlled clinical studies in 728 adult patients with moderately to severely active UC (Mayo score 6 to 12 [of possible range 0 to 12], Endoscopy subscore ≥2) with an inadequate response to conventional oral therapies (Studies UC I and UC II). Concomitant treatment with stable doses of aminosalicylates, corticosteroids and/or immunomodulatory agents was permitted. Corticosteroid taper was permitted after Week 8. Patients were randomized at Week 0 to receive either placebo, 5 mg/kg infliximab or 10 mg/kg infliximab at Weeks 0, 2, 6, and every 8 weeks thereafter through Week 46 in Study UC I, and at Weeks 0, 2, 6, and every 8 weeks thereafter through Week 22 in Study UC II. In Study UC II, patients were allowed to continue blinded therapy to Week 46 at the investigator's discretion.
Adult patients in Study UC I had failed to respond or were intolerant to oral corticosteroids, 6-MP, or AZA. Adult patients in Study UC II had failed to respond or were intolerant to the above treatments and/or aminosalicylates. Similar proportions of patients in Studies UC I and UC II were receiving corticosteroids (61% and 51%, respectively), 6-MP/AZA (49% and 43%) and aminosalicylates (70% and 75%) at baseline. More patients in Study UC II than UC I were taking solely aminosalicylates for UC (26% vs. 11%, respectively). Clinical response was defined as a decrease from baseline in the Mayo score by ≥30% and ≥3 points, accompanied by a decrease in the rectal bleeding subscore of ≥1 or a rectal bleeding subscore of 0 or 1.
Clinical Response, Clinical Remission, and Mucosal Healing
In both Study UC I and Study UC II, greater percentages of patients in both infliximab groups achieved clinical response, clinical remission and mucosal healing than in the placebo group. Each of these effects was maintained through the end of each trial (Week 54 in Study UC I, and Week 30 in Study UC II). In addition, a greater proportion of patients in infliximab groups demonstrated sustained response and sustained remission than in the placebo groups (Table 5).
Of patients on corticosteroids at baseline, greater proportions of adult patients in groups treated with infliximab were in clinical remission and able to discontinue corticosteroids at Week 30 compared with the patients in the placebo treatment groups (22% in infliximab treatment groups vs. 10% in placebo group in Study UC I; 23% in infliximab treatment groups vs. 3% in placebo group in Study UC II). In Study UC I, this effect was maintained through Week 54 (21% in infliximab treatment groups vs. 9% in placebo group). The infliximab-associated response was generally similar in the 5 mg/kg and 10 mg/kg dose groups.
| Study UC I | Study UC II | |||||
|---|---|---|---|---|---|---|
| Placebo | 5 mg/kg Infliximab | 10 mg/kg Infliximab | Placebo | 5 mg/kg Infliximab | 10 mg/kg Infliximab | |
| ||||||
Patients randomized | 121 | 121 | 122 | 123 | 121 | 120 |
Week 8 | 37% | 69%‡ | 62%‡ | 29% | 65%‡ | 69%‡ |
Week 30 | 30% | 52%‡ | 51%§ | 26% | 47%‡ | 60%‡ |
Week 54 | 20% | 45%‡ | 44%‡ | NA | NA | NA |
Sustained Response† | ||||||
(Clinical response at both Week 8 and 30) | 23% | 49%‡ | 46%‡ | 15% | 41%‡ | 53%‡ |
(Clinical response at Weeks 8, 30 and 54) | 14% | 39%‡ | 37%‡ | NA | NA | NA |
Week 8 | 15% | 39%‡ | 32%§ | 6% | 34%‡ | 28%‡ |
Week 30 | 16% | 34%§ | 37%‡ | 11% | 26%§ | 36%‡ |
Week 54 | 17% | 35%§ | 34%§ | NA | NA | NA |
Sustained Remission† | ||||||
(Clinical remission at both Week 8 and 30) | 8% | 23%§ | 26%‡ | 2% | 15%‡ | 23%‡ |
(Clinical remission at Weeks 8, 30 and 54) | 7% | 20%§ | 20%§ | NA | NA | NA |
Week 8 | 34% | 62%‡ | 59%‡ | 31% | 60%‡ | 62%‡ |
Week 30 | 25% | 50%‡ | 49%‡ | 30% | 46%§ | 57%‡ |
Week 54 | 18% | 45%‡ | 47%‡ | NA | NA | NA |
The improvement with infliximab was consistent across all Mayo subscores through Week 54 (Study UC I shown in Table 6; Study UC II through Week 30 was similar).
| Study UC I | |||
|---|---|---|---|
| Infliximab | |||
| Placebo (n=121) | 5 mg/kg (n=121) | 10 mg/kg (n=122) | |
Stool frequency | |||
Baseline | 17% | 17% | 10% |
Week 8 | 35% | 60% | 58% |
Week 30 | 35% | 51% | 53% |
Week 54 | 31% | 52% | 51% |
Rectal bleeding | |||
Baseline | 54% | 40% | 48% |
Week 8 | 74% | 86% | 80% |
Week 30 | 65% | 74% | 71% |
Week 54 | 62% | 69% | 67% |
Physician's Global Assessment | |||
Baseline | 4% | 6% | 3% |
Week 8 | 44% | 74% | 64% |
Week 30 | 36% | 57% | 55% |
Week 54 | 26% | 53% | 53% |
Endoscopy findings | |||
Baseline | 0% | 0% | 0% |
Week 8 | 34% | 62% | 59% |
Week 30 | 26% | 51% | 52% |
Week 54 | 21% | 50% | 51% |
14.4 Pediatric Ulcerative Colitis
The safety and effectiveness of infliximab products for reducing signs and symptoms and inducing and maintaining clinical remission in pediatric patients aged 6 years and older with moderately to severely active UC who have had an inadequate response to conventional therapy are supported by evidence from adequate and well-controlled studies of infliximab in adults. Additional safety and pharmacokinetic data were collected in an open-label pediatric UC trial in 60 pediatric patients aged 6 through 17 years (median age 14.5 years) with moderately to severely active UC (Mayo score of 6 to 12; Endoscopic subscore ≥2) and an inadequate response to conventional therapies. At baseline, the median Mayo score was 8, 53% of patients were receiving immunomodulator therapy (6-MP/AZA/MTX), and 62% of patients were receiving corticosteroids (median dose 0.5 mg/kg/day in prednisone equivalents). Discontinuation of immunomodulators and corticosteroid taper were permitted after Week 0.
All patients received induction dosing of 5 mg/kg infliximab at Weeks 0, 2, and 6. Patients who did not respond to infliximab at Week 8 received no further infliximab and returned for safety follow-up. At Week 8, 45 patients were randomized to a maintenance regimen of 5 mg/kg infliximab given either every 8 weeks through Week 46 or every 12 weeks through Week 42. Patients were allowed to change to a higher dose and/or more frequent administration schedule if they experienced loss of response.
Clinical response at Week 8 was defined as a decrease from baseline in the Mayo score by ≥30% and ≥3 points, including a decrease in the rectal bleeding subscore by ≥1 points or achievement of a rectal bleeding subscore of 0 or 1.
Clinical remission at Week 8 was measured by the Mayo score, defined as a Mayo score of ≤2 points with no individual subscore >1. Clinical remission was also assessed at Week 8 and Week 54 using the Pediatric Ulcerative Colitis Activity Index (PUCAI)1 score and was defined by a PUCAI score of <10 points.
Endoscopies were performed at baseline and at Week 8. A Mayo endoscopy subscore of 0 indicated normal or inactive disease and a subscore of 1 indicated mild disease (erythema, decreased vascular pattern, or mild friability).
Of the 60 patients treated, 44 were in clinical response at Week 8. Of 32 patients taking concomitant immunomodulators at baseline, 23 achieved clinical response at Week 8, compared to 21 of 28 of those not taking concomitant immunomodulators at baseline. At Week 8, 24 of 60 patients were in clinical remission as measured by the Mayo score and 17 of 51 patients were in remission as measured by the PUCAI score.
At Week 54, 8 of 21 patients in the every 8-week maintenance group and 4 of 22 patients in the every 12-week maintenance group achieved remission as measured by the PUCAI score.
During maintenance phase, 23 of 45 randomized patients (9 in the every 8-week group and 14 in the every 12-week group) required an increase in their dose and/or increase in frequency of infliximab administration due to loss of response. Nine of the 23 patients who required a change in dose had achieved remission at Week 54. Seven of those patients received the 10 mg/kg every 8-week dosing.
14.5 Rheumatoid Arthritis
The safety and efficacy of infliximab in adult patients with RA were assessed in 2 multicenter, randomized, double-blind, pivotal trials: ATTRACT (Study RA I) and ASPIRE (Study RA II). Concurrent use of stable doses of folic acid, oral corticosteroids (≤10 mg/day) and/or non-steroidal anti-inflammatory drugs (NSAIDs) was permitted.
Study RA I was a placebo-controlled study of 428 patients with active RA despite treatment with MTX. Patients enrolled had a median age of 54 years, median disease duration of 8.4 years, median swollen and tender joint count of 20 and 31 respectively, and were on a median dose of 15 mg/wk of MTX. Patients received either placebo+MTX or one of 4 doses/schedules of the infliximab+MTX: 3 mg/kg or 10 mg/kg infliximab by IV infusion at Weeks 0, 2 and 6 followed by additional infusions every 4 or 8 weeks in combination with MTX.
Study RA II was a placebo-controlled study of 3 active treatment arms in 1004 MTX naïve patients of 3 or fewer years' duration active RA. Patients enrolled had a median age of 51 years with a median disease duration of 0.6 years, median swollen and tender joint count of 19 and 31, respectively, and >80% of patients had baseline joint erosions. At randomization, all patients received MTX (optimized to 20 mg/wk by Week 8) and either placebo, 3 mg/kg or 6 mg/kg of infliximab at Weeks 0, 2, and 6 and every 8 weeks thereafter.
Data on use of infliximab products without concurrent MTX are limited [see Adverse Reactions (6.1)].
Clinical Response
In Study RA I, all doses/schedules of infliximab+MTX resulted in improvement in signs and symptoms as measured by the American College of Rheumatology response criteria (ACR 20) with a higher percentage of patients achieving an ACR 20, 50 and 70 compared to placebo+MTX (Table 7). This improvement was observed at Week 2 and maintained through Week 102. Greater effects on each component of the ACR 20 were observed in all patients treated with infliximab+MTX compared to placebo+MTX (Table 8). More patients treated with infliximab reached a major clinical response than placebo-treated patients (Table 7).
In Study RA II, after 54 weeks of treatment, both doses of infliximab+MTX resulted in statistically significantly greater response in signs and symptoms compared to MTX alone as measured by the proportion of patients achieving ACR 20, 50 and 70 responses (Table 7). More patients treated with infliximab reached a major clinical response than placebo-treated patients (Table 7).
Study RA I | Study RA II | ||||||||
Infliximab+MTX | Infliximab+MTX | ||||||||
3 mg/kg | 10 mg/kg | 3 mg/kg | 6 mg/kg | ||||||
Response | Placebo +MTX | q8 wks | q4 wks | q8 wks | q4 wks | Placebo +MTX | q8 wks | q8 wks | |
ACR 20 | |||||||||
Week 30 | 20% | 50%* | 50% * | 52% * | 58% * | N/A | N/A | N/A | |
Week 54 | 17% | 42% * | 48% * | 59% * | 59% * | 54% | 62% † | 66% * | |
ACR 50 | |||||||||
Week 30 | 5% | 27% * | 29% * | 31% * | 26% * | N/A | N/A | N/A | |
Week 54 | 9% | 21% † | 34% * | 40% * | 38% * | 32% | 46% * | 50% * | |
ACR 70 | |||||||||
Week 30 | 0% | 8%‡ | 11% ‡ | 18% * | 11% * | N/A | N/A | N/A | |
Week 54 | 2% | 11% † | 18% * | 26% * | 19% * | 21% | 33% ‡ | 37% * | |
Major clinical response§ | 0% | 7% † | 8% ‡ | 15% * | 6% † | 8% | 12% | 17% * | |
| Placebo+MTX (n=88) | Infliximab+MTX* (n=340) | |||
|---|---|---|---|---|
| Parameter (medians) | Baseline | Week 54 | Baseline | Week 54 |
No. of Tender Joints | 24 | 16 | 32 | 8 |
No. of Swollen Joints | 19 | 13 | 20 | 7 |
Pain† | 6.7 | 6.1 | 6.8 | 3.3 |
Physician's Global Assessment† | 6.5 | 5.2 | 6.2 | 2.1 |
Patient's Global Assessment† | 6.2 | 6.2 | 6.3 | 3.2 |
Disability Index (HAQ-DI)‡ | 1.8 | 1.5 | 1.8 | 1.3 |
CRP (mg/dL) | 3.0 | 2.3 | 2.4 | 0.6 |
Radiographic Response
Structural damage in both hands and feet was assessed radiographically at Week 54 by the change from baseline in the van der Heijde-modified Sharp (vdH-S) score, a composite score of structural damage that measures the number and size of joint erosions and the degree of joint space narrowing in hands/wrists and feet.
In Study RA I, approximately 80% of patients had paired X-ray data at 54 weeks and approximately 70% at 102 weeks. The inhibition of progression of structural damage was observed at 54 weeks (Table 9) and maintained through 102 weeks.
In Study RA II, >90% of patients had at least 2 evaluable X-rays. Inhibition of progression of structural damage was observed at Weeks 30 and 54 (Table 9) in infliximab+MTX groups compared to MTX alone. Patients treated with infliximab+MTX demonstrated less progression of structural damage compared to MTX alone, whether baseline acute-phase reactants (ESR and CRP) were normal or elevated: patients with elevated baseline acute-phase reactants treated with MTX alone demonstrated a mean progression in vdH-S score of 4.2 units compared to patients treated with infliximab+MTX who demonstrated 0.5 units of progression; patients with normal baseline acute phase reactants treated with MTX alone demonstrated a mean progression in vdH-S score of 1.8 units compared to infliximab+MTX who demonstrated 0.2 units of progression. Of patients receiving infliximab+MTX, 59% had no progression (vdH-S score ≤0 unit) of structural damage compared to 45% of patients receiving MTX alone. In a subset of patients who began the study without erosions, infliximab+MTX maintained an erosion-free state at 1 year in a greater proportion of patients than MTX alone, 79% (77/98) vs. 58% (23/40), respectively (P<0.01). Fewer patients in infliximab+MTX groups (47%) developed erosions in uninvolved joints compared to MTX alone (59%).
| Study RA I | Study RA II | ||||||
|---|---|---|---|---|---|---|---|
| Infliximab+MTX | Infliximab+MTX | ||||||
| 3 mg/kg | 10 mg/kg | 3 mg/kg | 6 mg/kg | ||||
| Placebo + MTX (n=64) | q8 wks (n=71) | q8 wks (n=77) | Placebo + MTX (n=282) | q8 wks (n=359) | q8 wks (n=363) | ||
| |||||||
Total Score | |||||||
Baseline | |||||||
Mean | 79 | 78 | 65 | 11.3 | 11.6 | 11.2 | |
Median | 55 | 57 | 56 | 5.1 | 5.2 | 5.3 | |
Change from baseline | |||||||
Mean | 6.9 | 1.3* | 0.2* | 3.7 | 0.4* | 0.5* | |
Median | 4.0 | 0.5 | 0.5 | 0.4 | 0.0 | 0.0 | |
Erosion Score | |||||||
Baseline | |||||||
Mean | 44 | 44 | 33 | 8.3 | 8.8 | 8.3 | |
Median | 25 | 29 | 22 | 3.0 | 3.8 | 3.8 | |
Change from baseline | |||||||
Mean | 4.1 | 0.2* | 0.2* | 3.0 | 0.3* | 0.1* | |
Median | 2.0 | 0.0 | 0.5 | 0.3 | 0.0 | 0.0 | |
JSN Score | |||||||
Baseline | |||||||
Mean | 36 | 34 | 31 | 3.0 | 2.9 | 2.9 | |
Median | 26 | 29 | 24 | 1.0 | 1.0 | 1.0 | |
Change from baseline | |||||||
Mean | 2.9 | 1.1* | 0.0* | 0.6 | 0.1* | 0.2 | |
Median | 1.5 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | |
Physical Function Response
Physical function and disability were assessed using the Health Assessment Questionnaire (HAQ-DI) and the general health-related quality of life questionnaire SF-36.
In Study RA I, all doses/schedules of infliximab+MTX showed significantly greater improvement from baseline in HAQ-DI and SF-36 physical component summary score averaged over time through Week 54 compared to placebo+MTX, and no worsening in the SF-36 mental component summary score. The median (interquartile range) improvement from baseline to Week 54 in HAQ-DI was 0.1 (-0.1, 0.5) for the placebo+MTX group and 0.4 (0.1, 0.9) for infliximab+MTX (p<0.001). Both HAQ-DI and SF-36 effects were maintained through Week 102. Approximately 80% of patients in all doses/schedules of infliximab+MTX remained in the trial through 102 weeks.
In Study RA II, both treatment groups of infliximab showed greater improvement in HAQ-DI from baseline averaged over time through Week 54 compared to MTX alone; 0.7 for infliximab+MTX vs. 0.6 for MTX alone (P≤0.001). No worsening in the SF-36 mental component summary score was observed.
14.6 Ankylosing Spondylitis
The safety and efficacy of infliximab were assessed in a randomized, multicenter, double-blind, placebo-controlled study in 279 adult patients with active AS. Patients were between 18 and 74 years of age, and had AS as defined by the modified New York criteria for Ankylosing Spondylitis. Patients were to have had active disease as evidenced by both a Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) score >4 (possible range 0–10) and spinal pain >4 (on a Visual Analog Scale [VAS] of 0–10). Patients with complete ankylosis of the spine were excluded from study participation, and the use of Disease Modifying Anti-Rheumatic Drugs (DMARDs) and systemic corticosteroids were prohibited. Doses of 5 mg/kg of infliximab or placebo were administered intravenously at Weeks 0, 2, 6, 12 and 18.
At 24 weeks, improvement in the signs and symptoms of AS, as measured by the proportion of patients achieving a 20% improvement in ASAS response criteria (ASAS 20), was seen in 60% of patients in the infliximab-treated group vs. 18% of patients in the placebo group (p<0.001). Improvement was observed at Week 2 and maintained through Week 24 (Figure 3 and Table 10).
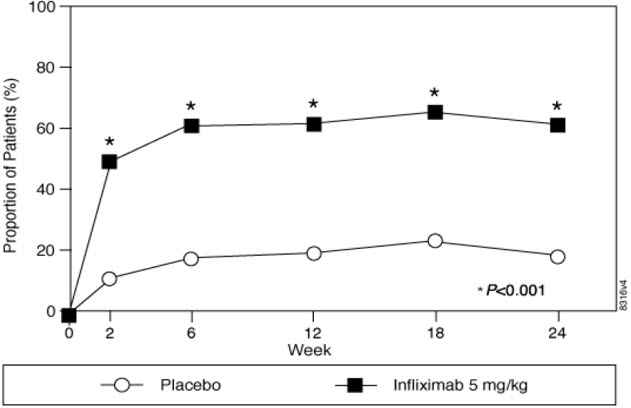
Figure 3 Proportion of Adult AS Patients Who Achieved ASAS 20 Response
At 24 weeks, the proportions of patients achieving a 50% and a 70% improvement in the signs and symptoms of AS, as measured by ASAS response criteria (ASAS 50 and ASAS 70, respectively), were 44% and 28%, respectively, for patients receiving infliximab, compared to 9% and 4%, respectively, for patients receiving placebo (P<0.001, infliximab vs. placebo). A low level of disease activity (defined as a value <20 [on a scale of 0 – 100 mm] in each of the 4 ASAS response parameters) was achieved in 22% of patients treated with infliximab vs. 1% in placebo-treated patients (P<0.001).
| Placebo (n=78) | Infliximab 5 mg/kg (n=201) | ||||
|---|---|---|---|---|---|
| Baseline | 24 Weeks | Baseline | 24 Weeks | P-value | |
| |||||
ASAS 20 response | |||||
Criteria (Mean) | |||||
Patient Global Assessment* | 6.6 | 6.0 | 6.8 | 3.8 | <0.001 |
Spinal pain* | 7.3 | 6.5 | 7.6 | 4.0 | <0.001 |
BASFI† | 5.8 | 5.6 | 5.7 | 3.6 | <0.001 |
Inflammation‡ | 6.9 | 5.8 | 6.9 | 3.4 | <0.001 |
Acute Phase Reactants | |||||
Median CRP§ (mg/dL) | 1.7 | 1.5 | 1.5 | 0.4 | <0.001 |
Spinal Mobility (cm, Mean) | |||||
Modified Schober's test¶ | 4.0 | 5.0 | 4.3 | 4.4 | 0.75 |
Chest expansion¶ | 3.6 | 3.7 | 3.3 | 3.9 | 0.04 |
Tragus to wall¶ | 17.3 | 17.4 | 16.9 | 15.7 | 0.02 |
Lateral spinal flexion¶ | 10.6 | 11.0 | 11.4 | 12.9 | 0.03 |
The median improvement from baseline in the general health-related quality-of-life questionnaire SF-36 physical component summary score at Week 24 was 10.2 for the infliximab group vs. 0.8 for the placebo group (P<0.001). There was no change in the SF-36 mental component summary score in either the infliximab group or the placebo group.
Results of this study were similar to those seen in a multicenter double-blind, placebo-controlled study of 70 patients with AS.
14.7 Psoriatic Arthritis
Safety and efficacy of infliximab were assessed in a multicenter, double-blind, placebo-controlled study in 200 adult patients with active PsA despite DMARD or NSAID therapy (≥5 swollen joints and ≥5 tender joints) with 1 or more of the following subtypes: arthritis involving DIP joints (n=49), arthritis mutilans (n=3), asymmetric peripheral arthritis (n=40), polyarticular arthritis (n=100), and spondylitis with peripheral arthritis (n=8). Patients also had Ps with a qualifying target lesion ≥2 cm in diameter. Forty-six percent of patients continued on stable doses of methotrexate (≤25 mg/week). During the 24-week double-blind phase, patients received either 5 mg/kg infliximab or placebo at Weeks 0, 2, 6, 14, and 22 (100 patients in each group). At Week 16, placebo patients with <10% improvement from baseline in both swollen and tender joint counts were switched to infliximab induction (early escape). At Week 24, all placebo-treated patients crossed over to infliximab induction. Dosing continued for all patients through Week 46.
Clinical Response
Treatment with infliximab resulted in improvement in signs and symptoms, as assessed by the ACR criteria, with 58% of patients treated with infliximab achieving ACR 20 at Week 14, compared with 11% of placebo-treated patients (P<0.001). The response was similar regardless of concomitant use of methotrexate. Improvement was observed as early as Week 2. At 6 months, the ACR 20/50/70 responses were achieved by 54%, 41%, and 27%, respectively, of patients receiving infliximab compared to 16%, 4%, and 2%, respectively, of patients receiving placebo. Similar responses were seen in patients with each of the subtypes of PsA, although few patients were enrolled with the arthritis mutilans and spondylitis with peripheral arthritis subtypes.
Compared to placebo, treatment with infliximab resulted in improvements in the components of the ACR response criteria, as well as in dactylitis and enthesopathy (Table 11). The clinical response was maintained through Week 54. Similar ACR responses were observed in an earlier randomized, placebo-controlled study of 104 PsA patients, and the responses were maintained through 98 weeks in an open-label extension phase.
| Patients Randomized | Placebo (n=100) | Infliximab 5 mg/kg* (n=100) | ||
|---|---|---|---|---|
| Baseline | Week 24 | Baseline | Week 24 | |
| ||||
Parameter (medians) | ||||
No. of Tender Joints† | 24 | 20 | 20 | 6 |
No. of Swollen Joints‡ | 12 | 9 | 12 | 3 |
Pain§ | 6.4 | 5.6 | 5.9 | 2.6 |
Physician's Global Assessment§ | 6.0 | 4.5 | 5.6 | 1.5 |
Patient's Global Assessment§ | 6.1 | 5.0 | 5.9 | 2.5 |
Disability Index (HAQ-DI)¶ | 1.1 | 1.1 | 1.1 | 0.5 |
CRP (mg/dL)# | 1.2 | 0.9 | 1.0 | 0.4 |
% Patients with 1 or more digits with dactylitis | 41 | 33 | 40 | 15 |
% Patients with enthesopathy | 35 | 36 | 42 | 22 |
Improvement in Psoriasis Area and Severity Index (PASI) in PsA patients with baseline body surface area (BSA) ≥3% (n=87 placebo, n=83 infliximab) was achieved at Week 14, regardless of concomitant methotrexate use, with 64% of patients treated with infliximab achieving at least 75% improvement from baseline vs. 2% of placebo-treated patients; improvement was observed in some patients as early as Week 2. At 6 months, the PASI 75 and PASI 90 responses were achieved by 60% and 39%, respectively, of patients receiving infliximab compared to 1% and 0%, respectively, of patients receiving placebo. The PASI response was generally maintained through Week 54. [See Clinical Studies (14.8)].
Radiographic Response
Structural damage in both hands and feet was assessed radiographically by the change from baseline in the van der Heijde-Sharp (vdH-S) score, modified by the addition of hand DIP joints. The total modified vdH-S score is a composite score of structural damage that measures the number and size of joint erosions and the degree of joint space narrowing (JSN) in the hands and feet. At Week 24, patients treated with infliximab had less radiographic progression than placebo-treated patients (mean change of -0.70 vs. 0.82, P<0.001). Patients treated with infliximab also had less progression in their erosion scores (-0.56 vs 0.51) and JSN scores (-0.14 vs 0.31). The patients in the infliximab group demonstrated continued inhibition of structural damage at Week 54. Most patients showed little or no change in the vdH-S score during this 12-month study (median change of 0 in both patients who initially received infliximab or placebo). More patients in the placebo group (12%) had readily apparent radiographic progression compared with the infliximab group (3%).
Physical Function
Physical function status was assessed using the HAQ Disability Index (HAQ-DI) and the SF-36 Health Survey. Patients treated with infliximab demonstrated significant improvement in physical function as assessed by HAQ-DI (median percent improvement in HAQ-DI score from baseline to Week 14 and 24 of 43% for infliximab-treated patients vs 0% for placebo-treated patients).
During the placebo-controlled portion of the trial (24 weeks), 54% of patients treated with infliximab achieved a clinically meaningful improvement in HAQ-DI (≥0.3 unit decrease) compared to 22% of placebo-treated patients. Patients treated with infliximab also demonstrated greater improvement in the SF-36 physical and mental component summary scores than placebo-treated patients. The responses were maintained for up to 2 years in an open-label extension study.
14.8 Plaque Psoriasis
The safety and efficacy of infliximab were assessed in 3 randomized, double-blind, placebo-controlled studies in patients 18 years of age and older with chronic, stable Ps involving ≥10% BSA, a minimum PASI score of 12, and who were candidates for systemic therapy or phototherapy. Patients with guttate, pustular, or erythrodermic psoriasis were excluded from these studies. No concomitant anti-psoriatic therapies were allowed during the study, with the exception of low-potency topical corticosteroids on the face and groin after Week 10 of study initiation.
Study I (EXPRESS) evaluated 378 patients who received placebo or infliximab at a dose of 5 mg/kg at Weeks 0, 2, and 6 (induction therapy), followed by maintenance therapy every 8 weeks. At Week 24, the placebo group crossed over to infliximab induction therapy (5 mg/kg), followed by maintenance therapy every 8 weeks. Patients originally randomized to infliximab continued to receive infliximab 5 mg/kg every 8 weeks through Week 46. Across all treatment groups, the median baseline PASI score was 21 and the baseline Static Physician Global Assessment (sPGA) score ranged from moderate (52% of patients) to marked (36%) to severe (2%). In addition, 75% of patients had a BSA >20%. Seventy-one percent of patients previously received systemic therapy, and 82% received phototherapy.
Study II (EXPRESS II) evaluated 835 patients who received placebo or infliximab at doses of 3 mg/kg or 5 mg/kg at Weeks 0, 2, and 6 (induction therapy). At Week 14, within each infliximab dose group, patients were randomized to either scheduled (every 8 weeks) or as needed (PRN) maintenance treatment through Week 46. At Week 16, the placebo group crossed over to infliximab induction therapy (5 mg/kg), followed by maintenance therapy every 8 weeks. Across all treatment groups, the median baseline PASI score was 18, and 63% of patients had a BSA >20%. Fifty-five percent of patients previously received systemic therapy, and 64% received a phototherapy.
Study III (SPIRIT) evaluated 249 patients who had previously received either psoralen plus ultraviolet A treatment (PUVA) or other systemic therapy for their psoriasis. These patients were randomized to receive either placebo or infliximab at doses of 3 mg/kg or 5 mg/kg at Weeks 0, 2, and 6. At Week 26, patients with a sPGA score of moderate or worse (greater than or equal to 3 on a scale of 0 to 5) received an additional dose of the randomized treatment. Across all treatment groups, the median baseline PASI score was 19, and the baseline sPGA score ranged from moderate (62% of patients) to marked (22%) to severe (3%). In addition, 75% of patients had a BSA >20%. Of the enrolled patients, 114 (46%) received the Week 26 additional dose.
In Studies I, II and III, the primary endpoint was the proportion of patients who achieved a reduction in score of at least 75% from baseline at Week 10 by the PASI (PASI 75). In Study I and Study III, another evaluated outcome included the proportion of patients who achieved a score of "cleared" or "minimal" by the sPGA. The sPGA is a 6-category scale ranging from "5 = severe" to "0 = cleared" indicating the physician's overall assessment of the psoriasis severity focusing on induration, erythema, and scaling. Treatment success, defined as "cleared" or "minimal," consisted of none or minimal elevation in plaque, up to faint red coloration in erythema, and none or minimal fine scale over <5% of the plaque.
Study II also evaluated the proportion of patients who achieved a score of "clear" or "excellent" by the relative Physician's Global Assessment (rPGA). The rPGA is a 6-category scale ranging from "6 = worse" to "1 = clear" that was assessed relative to baseline. Overall lesions were graded with consideration to the percent of body involvement as well as overall induration, scaling, and erythema. Treatment success, defined as "clear" or "excellent," consisted of some residual pinkness or pigmentation to marked improvement (nearly normal skin texture; some erythema may be present). The results of these studies are presented in Table 12.
| Placebo | Infliximab | ||
|---|---|---|---|
| 3 mg/kg | 5 mg/kg | ||
Psoriasis Study I - patients randomized* | 77 | --- | 301 |
PASI 75 | 2 (3%) | --- | 242 (80%)† |
sPGA | 3 (4%) | --- | 242 (80%)† |
Psoriasis Study II - patients randomized* | 208 | 313 | 314 |
PASI 75 | 4 (2%) | 220 (70%)† | 237 (75%)† |
rPGA | 2 (1%) | 217 (69%)† | 234 (75%)† |
Psoriasis Study III - patients randomized‡ | 51 | 99 | 99 |
PASI 75 | 3 (6%) | 71 (72%)† | 87 (88%)† |
sPGA | 5 (10%) | 71 (72%)† | 89 (90%)† |
In Study I, in the subgroup of patients with more extensive Ps who had previously received phototherapy, 85% of patients on 5 mg/kg infliximab achieved a PASI 75 at Week 10 compared with 4% of patients on placebo.
In Study II, in the subgroup of patients with more extensive Ps who had previously received phototherapy, 72% and 77% of patients on 3 mg/kg and 5 mg/kg infliximab achieved a PASI 75 at Week 10 respectively compared with 1% on placebo. In Study II, among patients with more extensive Ps who had failed or were intolerant to phototherapy, 70% and 78% of patients on 3 mg/kg and 5 mg/kg infliximab achieved a PASI 75 at Week 10 respectively, compared with 2% on placebo.
Maintenance of response was studied in a subset of 292 and 297 patients treated with infliximab in the 3 mg/kg and 5 mg/kg groups; respectively, in Study II. Stratified by PASI response at Week 10 and investigational site, patients in the active treatment groups were re-randomized to either a scheduled or as needed maintenance (PRN) therapy, beginning on Week 14.
The groups that received a maintenance dose every 8 weeks appear to have a greater percentage of patients maintaining a PASI 75 through week 50 as compared to patients who received the as-needed or PRN doses, and the best response was maintained with the 5 mg/kg every 8-week dose. These results are shown in Figure 4. At Week 46, when infliximab serum concentrations were at trough level, in the every 8-week dose group, 54% of patients in the 5 mg/kg group compared to 36% in the 3 mg/kg group achieved PASI 75. The lower percentage of PASI 75 responders in the 3 mg/kg every 8-week dose group compared to the 5 mg/kg group was associated with a lower percentage of patients with detectable trough serum infliximab levels.
This may be related in part to higher antibody rates [see Adverse Reactions (6.1)]. In addition, in a subset of patients who had achieved a response at Week 10, maintenance of response appears to be greater in patients who received infliximab every 8 weeks at the 5 mg/kg dose. Regardless of whether the maintenance doses are PRN or every 8 weeks, there is a decline in response in a subpopulation of patients in each group over time. The results of Study I through Week 50 in the 5 mg/kg every 8 weeks maintenance dose group were similar to the results from Study II.
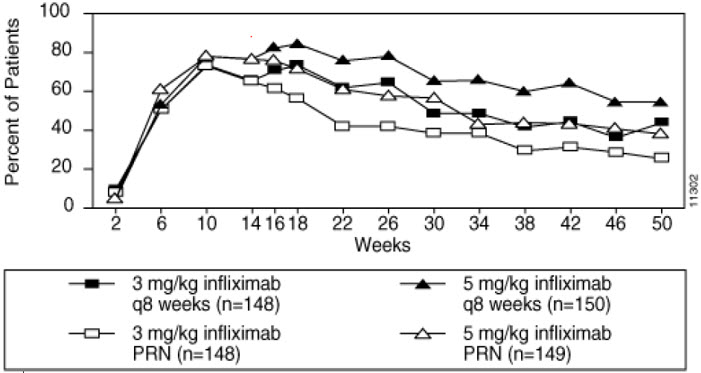
Figure 4 Proportion of Adult Ps Patients Who Achieved ≥75% Improvement in PASI from Baseline through Week 50 (patients randomized at Week 14)
Efficacy and safety of infliximab treatment beyond 50 weeks have not been evaluated in patients with Ps.
MEDICATION GUIDE
| This Medication Guide has been approved by the U.S. Food and Drug Administration Revised: September 2025 | |||
MEDICATION GUIDE | |||
Read the Medication Guide that comes with INFLECTRA before you receive the first treatment, and before each time you get a treatment of INFLECTRA. This Medication Guide does not take the place of talking with your doctor about your medical condition or treatment.
Before starting INFLECTRA, tell your doctor if you:
After starting INFLECTRA, if you have an infection, any sign of an infection including a fever, cough, flu-like symptoms, or have open cuts or sores on your body, call your doctor right away. INFLECTRA can make you more likely to get infections or make any infection that you have worse.
See the section "What are the possible side effects of INFLECTRA?" below for more information. | |||
What is INFLECTRA?
INFLECTRA blocks the action of a protein in your body called tumor necrosis factor-alpha (TNF-alpha). TNF-alpha is made by your body's immune system. People with certain diseases have too much TNF-alpha that can cause the immune system to attack normal healthy parts of the body. INFLECTRA can block the damage caused by too much TNF-alpha. | |||
Who should not receive INFLECTRA?
| |||
What should I tell my doctor before starting treatment with INFLECTRA?
If you have a baby and you were receiving INFLECTRA during your pregnancy, it is important to tell your baby's doctor and other healthcare professionals about your INFLECTRA use so they can decide when your baby should receive any vaccine. Certain vaccinations can cause infections. | |||
How should I receive INFLECTRA?
| |||
What should I avoid while receiving INFLECTRA? | |||
What are the possible side effects of INFLECTRA?
| |||
| |||
| |||
| |||
Heart Failure | |||
|
| ||
Treatment with INFLECTRA may need to be stopped if you get new or worse congestive heart failure. | |||
|
| ||
Blood Problems | |||
|
| ||
Nervous System Disorders | |||
|
| ||
Some patients have experienced a stroke within approximately 24 hours of their infusion with infliximab products. Tell your doctor right away if you have symptoms of a stroke which may include: numbness or weakness of the face, arm or leg, especially on one side of the body; sudden confusion, trouble speaking or understanding; sudden trouble seeing in one or both eyes, sudden trouble walking, dizziness, loss of balance or coordination or a sudden, severe headache. | |||
|
| ||
Some patients treated with infliximab products have had delayed allergic reactions. The delayed reactions occurred within 3 to 12 days after receiving treatment with infliximab products. Tell your doctor right away if you have any of these signs of delayed allergic reaction to INFLECTRA: | |||
|
| ||
Lupus-like Syndrome | |||
|
| ||
Psoriasis | |||
|
| ||
Infusion reactions can happen up to 2 hours after your infusion of INFLECTRA. Symptoms of infusion reactions may include: | |||
|
|
| |
Children with Crohn's disease who received infliximab showed some differences in side effects of treatment compared with adults with Crohn's disease. The side effects that happened more in children were: anemia (low red blood cells), leukopenia (low white blood cells), flushing (redness or blushing), viral infections, neutropenia (low neutrophils, the white blood cells that fight infection), bone fracture, bacterial infection and allergic reactions of the breathing tract. Among patients who received infliximab for ulcerative colitis in clinical studies, more children had infections as compared with adults. | |||
General information about INFLECTRA | |||
What are the ingredients in INFLECTRA? | |||
Manufactured by: CELLTRION, Inc. 23, Academy-ro, Yeonsu-gu, Incheon, 22014, Republic of Korea |  | ||
Additional Resources
Chat online with Pfizer Medical Information regarding your inquiry on a Pfizer medicine or vaccine.
Speak with a Pfizer Medical Information Professional regarding your Pfizer medicine or vaccine inquiry.
Available 9AM-5PM ET Monday to Friday; excluding holidays.
Submit a medical question for a Pfizer medicine or a vaccine.
The submission will be reviewed during our standard business hours.
To report an adverse event related to a Pfizer product and you are not part of a clinical trial* for this medication, click the link below to submit your information:
Pfizer Safety Reporting Site
*If you are involved in a clinical trial for either product, adverse events should be reported to your coordinating study site.
If you cannot use the above website to report an adverse event related to a Pfizer medication, please call (800) 438-1985.
You may also contact the U.S. Food and Drug Administration (FDA) directly to report adverse events or product quality concerns either online at www.fda.gov/medwatch or by calling (800) 332-1088.