(palbociclib)
14 CLINICAL STUDIES
14 CLINICAL STUDIES
14.1 HER2-Negative Metastatic Breast Cancer
PALOMA-2: IBRANCE plus Letrozole
Patients with ER-positive, HER2-negative advanced or metastatic breast cancer for initial endocrine-based therapy
PALOMA-2 was an international, randomized, double-blind, parallel-group, multicenter study of IBRANCE plus letrozole versus placebo plus letrozole conducted in postmenopausal women with ER-positive, HER2-negative advanced breast cancer who had not received previous systemic treatment for their advanced disease. A total of 666 patients were randomized 2:1 to IBRANCE plus letrozole or placebo plus letrozole. Randomization was stratified by disease site (visceral versus non-visceral), disease-free interval (de novo metastatic versus ≤12 months from the end of adjuvant treatment to disease recurrence versus >12 months from the end of adjuvant treatment to disease recurrence), and nature of prior (neo)adjuvant anticancer therapies (prior hormonal therapies versus no prior hormonal therapy). IBRANCE was given orally at a dose of 125 mg daily for 21 consecutive days followed by 7 days off treatment. Patients received study treatment until objective disease progression, symptomatic deterioration, unacceptable toxicity, death, or withdrawal of consent, whichever occurred first. The major efficacy outcome of the study was investigator-assessed progression-free survival (PFS) evaluated according to Response Evaluation Criteria in Solid Tumors Version 1.1 (RECIST). Additional efficacy outcome measures were confirmed overall response rate (ORR) as assessed by the investigator according to RECIST Version 1.1 and overall survival (OS).
Patients enrolled in this study had a median age of 62 years (range 28 to 89). The majority of patients were White (78%), and most patients had an Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 0 or 1 (98%). Forty-eight percent of patients had received chemotherapy and 56% had received antihormonal therapy in the neoadjuvant or adjuvant setting prior to their diagnosis of advanced breast cancer. Thirty-seven percent of patients had no prior systemic therapy in the neoadjuvant or adjuvant setting. The majority of patients (97%) had metastatic disease. Twenty-three percent of patients had bone only disease, and 49% of patients had visceral disease.
Major efficacy results from PALOMA-2 are summarized in Table 12 and Figure 1. Consistent results were observed across patient subgroups of disease-free interval (DFI), disease site, and prior therapy. The treatment effect of the combination on PFS was also supported by an independent review of radiographs. Based on the prespecified final OS analysis conducted after 435 events, OS was not statistically significant.
| IBRANCE plus Letrozole | Placebo plus Letrozole | |
|---|---|---|
| CI=confidence interval; ITT=Intent-to-Treat; N=number of patients; NE=not estimable; OS=overall survival; PFS=progression‑free survival. | ||
Progression-free survival for ITT (investigator assessment) | N=444 | N=222 |
Number of PFS events (%) | 194 (43.7) | 137 (61.7) |
Median progression-free survival | 24.8 (22.1, NE) | 14.5 (12.9, 17.1) |
Hazard ratio (95% CI) and p-value | ||
Objective Response for patients with measurable disease (investigator assessment) | N=338 | N=171 |
Objective response rate‡ (%, 95% CI) | 55.3 (49.9, 60.7) | 44.4 (36.9, 52.2) |
Overall survival for ITT | N=444 | N=222 |
Number of OS events (%) | 287 (64.6) | 148 (66.7) |
Median OS (months, 95% CI) | 53.8 (49.8, 59.2) | 49.8 (42.3, 56.4) |
Hazard ratio (95% CI) and p‑value | ||
Figure 1. Kaplan-Meier Plot of Progression-Free Survival – PALOMA-2 (Investigator Assessment, Intent-to-Treat Population) | ||
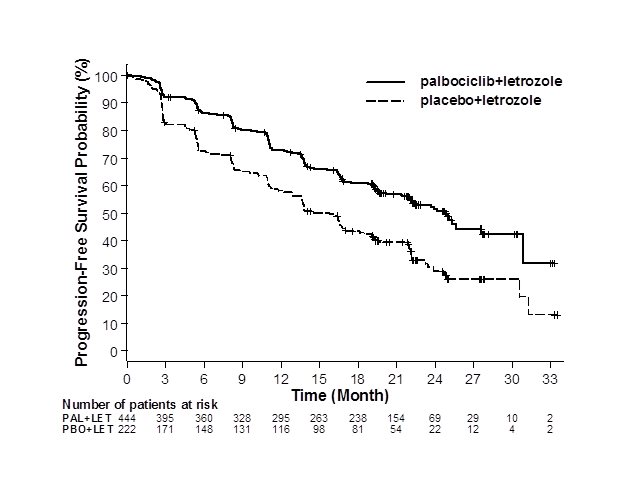 | ||
LET=letrozole; PAL=palbociclib; PBO=placebo. | ||
PALOMA-3: IBRANCE plus Fulvestrant
Patients with HR-positive, HER2-negative advanced or metastatic breast cancer who have had disease progression on or after prior adjuvant or metastatic endocrine therapy
PALOMA-3 was an international, randomized, double-blind, parallel-group, multicenter study of IBRANCE plus fulvestrant versus placebo plus fulvestrant conducted in women with HR-positive, HER2-negative advanced breast cancer, regardless of their menopausal status, whose disease progressed on or after prior endocrine therapy. A total of 521 pre/postmenopausal women were randomized 2:1 to IBRANCE plus fulvestrant or placebo plus fulvestrant and stratified by documented sensitivity to prior hormonal therapy, menopausal status at study entry (pre/peri versus postmenopausal), and presence of visceral metastases. IBRANCE was given orally at a dose of 125 mg daily for 21 consecutive days followed by 7 days off treatment. Pre/perimenopausal women were enrolled in the study and received the LHRH agonist goserelin for at least 4 weeks prior to and for the duration of PALOMA-3. Patients continued to receive assigned treatment until objective disease progression, symptomatic deterioration, unacceptable toxicity, death, or withdrawal of consent, whichever occurred first. The major efficacy outcome of the study was investigator-assessed PFS evaluated according to RECIST 1.1.
Patients enrolled in this study had a median age of 57 years (range 29 to 88). The majority of patients on study were White (74%), all patients had an ECOG PS of 0 or 1, and 80% were postmenopausal. All patients had received prior systemic therapy, and 75% of patients had received a previous chemotherapy regimen. Twenty-five percent of patients had received no prior therapy in the metastatic disease setting, 60% had visceral metastases, and 23% had bone only disease.
The results from the investigator-assessed PFS and final OS from PALOMA-3 are summarized in Table 13. The relevant Kaplan-Meier plots are shown in Figures 2 and 3, respectively. Consistent PFS results were observed across patient subgroups of disease site, sensitivity to prior hormonal therapy, and menopausal status. After a median follow-up time of 45 months, the final OS results were not statistically significant.
| CI=confidence interval; ITT=Intent-to-Treat; N=number of patients; OS=overall survival; PFS=progression-free survival. | ||
IBRANCE plus Fulvestrant | Placebo plus Fulvestrant | |
Progression-free survival for ITT | N=347 | N=174 |
Number of PFS events (%) | 145 (41.8) | 114 (65.5) |
Median PFS (months, 95% CI) | 9.5 (9.2, 11.0) | 4.6 (3.5, 5.6) |
Hazard ratio (95% CI) and p-value | 0.461 (0.360, 0.591), p<0.0001 | |
Objective Response for patients with measurable disease | N=267 | N=138 |
Objective response rate* (%, 95% CI) | 24.6 (19.6, 30.2) | 10.9 (6.2, 17.3) |
Overall survival for ITT | N=347 | N=174 |
Number of OS events (%) | 201 (57.9) | 109 (62.6) |
Median OS (months, 95% CI) | 34.9 (28.8, 40.0) | 28.0 (23.6, 34.6) |
Hazard ratio (95% CI) and p-value | ||
Figure 2. Kaplan-Meier Plot of Progression-Free Survival – PALOMA-3 (Investigator Assessment, Intent-to-Treat Population) | ||
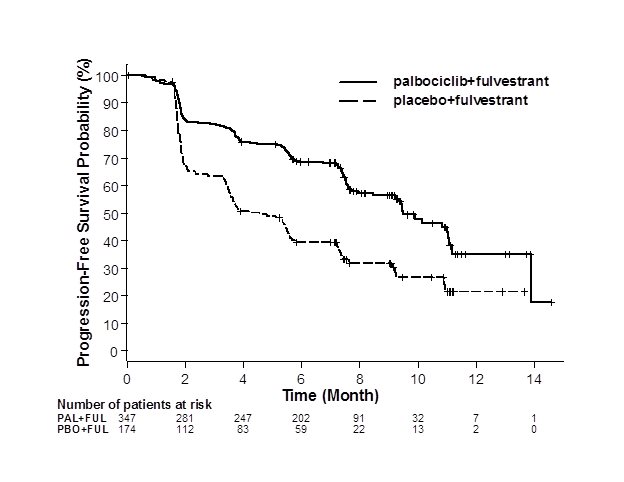 | ||
FUL=fulvestrant; PAL=palbociclib; PBO=placebo. | ||
Figure 3. Kaplan-Meier Plot of Overall Survival (Intent-to-Treat Population) – PALOMA-3 | ||
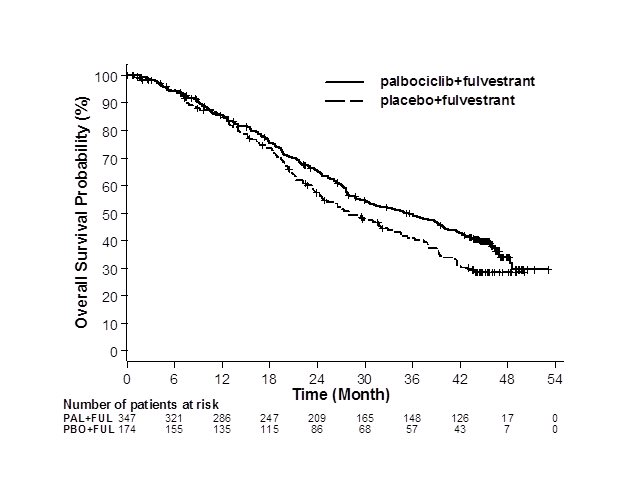 | ||
FUL=fulvestrant; PAL=palbociclib; PBO=placebo. | ||
INAVO120: IBRANCE plus Inavolisib and Fulvestrant
Adults with PIK3CA-mutated, HR-positive, HER2-negative, locally advanced or metastatic breast cancer whose disease progressed during or within 12 months of completing adjuvant endocrine therapy and who have not received prior systemic therapy for locally advanced or metastatic disease
INAVO120 (NCT04191499) was a randomized (1:1), double-blind, placebo-controlled trial evaluating the efficacy of inavolisib in combination with IBRANCE and fulvestrant in adult patients with endocrine-resistant PIK3CA-mutated, HR-positive, HER2-negative (defined as IHC 0 or 1+, or IHC 2+/ISH-), locally advanced or metastatic breast cancer whose disease progressed during or within 12 months of completing adjuvant endocrine therapy and who have not received prior systemic therapy for locally advanced or metastatic disease. Randomization was stratified by presence of visceral disease (yes or no), endocrine resistance (primary or secondary), and geographic region (North America/Western Europe, Asia, other).
Primary endocrine resistance was defined as relapse while on the first 2 years of adjuvant endocrine therapy (ET) and secondary endocrine resistance was defined as relapse while on adjuvant ET after at least 2 years or relapse within 12 months of completing adjuvant ET.
Patients were required to have a HbA1C <6% and fasting blood glucose <126 mg/dL. The study excluded patients with Type 1 diabetes mellitus or Type 2 diabetes mellitus requiring ongoing anti-hyperglycemic treatment at the start of study treatment.
PIK3CA mutation status was prospectively determined in a central laboratory using the FoundationOne® Liquid CDx assay on plasma-derived circulating tumor DNA (ctDNA) or in local laboratories using various validated polymerase chain reaction (PCR) or next-generation sequencing (NGS) assays on tumor tissue or plasma. All patients were required to provide both a freshly collected pre-treatment blood sample and a tumor tissue sample for central evaluation and determination of PIK3CA mutation(s) status.
Patients received either inavolisib 9 mg (n=161) or placebo (n=164) orally once daily, in combination with IBRANCE 125 mg orally once daily for 21 consecutive days followed by 7 days off treatment to comprise a cycle of 28 days, and fulvestrant 500 mg administered intramuscularly on Cycle 1, Days 1 and 15, and then on Day 1 of every 28-day cycle. Patients received treatment until disease progression or unacceptable toxicity. In addition, all pre/perimenopausal women and men received an LHRH agonist throughout therapy.
The baseline demographic and disease characteristics were: median age 54 years (range: 27 to 79 years); 98% female, of which 39% were pre/perimenopausal; 59% White, 38% Asian, 2.5% Unknown, 0.6% Black or African American; 6% Hispanic or Latino; and ECOG PS of 0 (63%) or 1 (36%). Tamoxifen (57%) and aromatase inhibitors (50%) were the most commonly used adjuvant endocrine therapies. Sixty-four percent of patients were considered to have secondary endocrine resistance. Eighty-three percent of patients had received prior chemotherapy (in the neo/adjuvant setting) and 1.2% of patients had been treated with a CDK4/6 inhibitor.
The major efficacy outcome measure was investigator (INV)-assessed PFS per RECIST version 1.1. Additional efficacy outcome measures included OS, INV-assessed ORR, and INV-assessed duration of response (DOR).
Efficacy results are summarized in Table 14 and Figure 4. INV-assessed PFS results were supported by consistent results from a blinded independent central review (BICR) assessment. At the time of the PFS analysis, OS data were not mature with 30% deaths in the overall population.
| CI=confidence interval; CR=complete response; DOR=duration of response; N=number of patients; PR=partial response. | ||
Efficacy Endpoint | IBRANCE plus Inavolisib and Fulvestrant N=161 | IBRANCE plus Placebo and Fulvestrant N=164 |
Patients with event (%) | 82 (51) | 113 (69) |
Median, months (95% CI) | 15.0 (11.3, 20.5) | 7.3 (5.6, 9.3) |
Hazard ratio (95% CI) | 0.43 (0.32, 0.59) | |
p-value | <0.0001 | |
Patients with CR or PR (%) | 94 (58) | 41 (25) |
95% CI | (50, 66) | (19, 32) |
Duration of Response† | ||
Median DOR, months (95% CI) | 18.4 (10.4, 22.2) | 9.6 (7.4, 16.6) |
Figure 4. Kaplan-Meier Curve for Investigator-Assessed Progression-Free Survival in INAVO120 |
 |
14.2 HER2-Positive Metastatic Breast Cancer
PATINA: IBRANCE in Combination with Trastuzumab, with or without Pertuzumab, and Endocrine Therapy (Fulvestrant or Aromatase inhibitors [Anastrozole, Letrozole or Exemestane])
Patients with HR-positive, HER2-positive locally advanced or metastatic breast cancer after induction therapy.
PATINA (NCT02947685) was a randomized (1:1), open-label trial evaluating the efficacy of IBRANCE in combination with trastuzumab, with or without pertuzumab, and endocrine therapy in adult patients with HR-positive, HER2-positive (defined as IHC 3+ or IHC 2+/ISH+) locally advanced or metastatic breast cancer who had no evidence of disease progression after induction treatment for their advanced disease.
Induction treatment was defined as 4-8 cycles of a taxane in combination with trastuzumab, with or without pertuzumab, received for the treatment of locally advanced or metastatic breast cancer in the first line setting and completed prior to randomization.
Patients were determined to have no evidence of disease progression if they had a CR, PR, or SD by local investigator assessment at the time of tumor assessment prior to randomization.
Randomization was stratified by pertuzumab use (yes vs. no), prior anti-HER2 therapy in the neo (adjuvant) setting (yes vs. no), type of endocrine therapy (aromatase inhibitor vs. fulvestrant) and best response to induction therapy (CR or PR vs. SD) determined by investigator assessment.
Patients received either IBRANCE in combination with trastuzumab, with or without pertuzumab, and endocrine therapy (n=261) or trastuzumab, with or without pertuzumab, and endocrine therapy alone (n=257). IBRANCE was given orally at a dose of 125 mg daily for 21 consecutive days followed by 7 days off treatment to comprise a cycle of 28 days. Patients received treatment until disease progression or unacceptable toxicity. All men and premenopausal women enrolled in the trial received an LHRH agonist when appropriate.
The major efficacy outcome measure was investigator-assessed PFS per RECIST version 1.1. Overall survival was an additional efficacy outcome.
The baseline demographic and disease characteristics were: median age 53 years (range 28 to 84 years); 99% female, of which 62% were postmenopausal; 77% White, 3% Black or African American, 2% Asian and 16% not specified; 14% Hispanic or Latino; and ECOG PS of 0 (57%) or 1 (43%). Aromatase inhibitors (91%) were the most commonly used endocrine therapy. Patients received a median of 6 cycles of induction chemotherapy in combination with trastuzumab, with or without pertuzumab, prior to study entry. Fifty percent had visceral metastases; 33% liver metastases, and 17% lung metastases.
Efficacy results are summarized in Table 15 and Figure 5. Median PFS cannot be adequately described because of censoring. At the time of the PFS analysis, OS data were not mature with 24% deaths in the overall population.
| CI=confidence interval; N=number of patients; PFS=progression-free survival. | ||
| ||
IBRANCE plus trastuzumab with or without pertuzumab and endocrine therapy (N=261) | Trastuzumab with or without pertuzumab and endocrine therapy (N=257) | |
Progression-free survival (investigator assessment) | ||
Patients with PFS event, n (%) | 124 (47.5) | 133 (51.8) |
Hazard ratio (95% CI) and p‑value* | 0.76 (0.59, 0.97), p=0.0134 | |
Figure 5. Kaplan-Meier Plot of Progression-Free Survival Based on Investigator Assessment - PATINA |
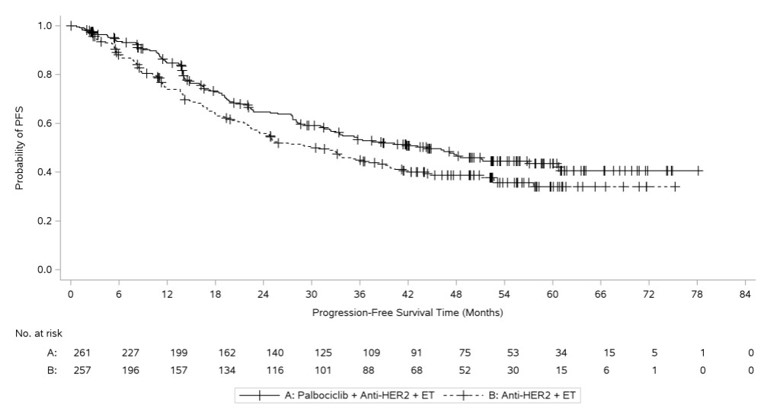 |
MEDICATION GUIDE
| This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: June 2026 | |||
PATIENT INFORMATION | |||
What is the most important information I should know about IBRANCE? IBRANCE may cause serious side effects, including:
If you develop low white blood cell counts during treatment with IBRANCE, your healthcare provider may stop your treatment, decrease your dose, or may tell you to wait to begin your treatment cycle. Tell your healthcare provider right away if you have signs and symptoms of low white blood cell counts or infections such as fever and chills.
Your healthcare provider may interrupt or stop treatment with IBRANCE completely if your symptoms are severe. See "What are the possible side effects of IBRANCE?" for more information about side effects. | |||
What is IBRANCE? IBRANCE is a prescription medicine used for: Human Epidermal Growth Factor Receptor 2 (HER2) – Negative Metastatic Breast Cancer
HER2-Positive Metastatic Breast Cancer
It is not known if IBRANCE is safe and effective in children. | |||
What should I tell my healthcare provider before taking IBRANCE? Before taking IBRANCE, tell your healthcare provider about all of your medical conditions, including if you:
Tell your healthcare provider about all of the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. IBRANCE and other medicines may affect each other causing side effects. Know the medicines you take. Keep a list of them to show your healthcare provider or pharmacist when you get a new medicine. | |||
How should I take IBRANCE?
| |||
What are the possible side effects of IBRANCE? IBRANCE may cause serious side effects. See "What is the most important information I should know about IBRANCE?" The most common side effects of IBRANCE when used with either letrozole or fulvestrant include:
| |||
|
| ||
|
| ||
The most common side effects of IBRANCE when used in combination with inavolisib plus fulvestrant include: | |||
|
| ||
The most common side effects of IBRANCE when used in combination with trastuzumab, with or without pertuzumab, and hormonal therapy include: | |||
|
| ||
IBRANCE may cause fertility problems in males. This may affect your ability to father a child. Talk to your healthcare provider about family planning options before starting IBRANCE if this is a concern for you. Tell your healthcare provider if you have any side effect that bothers you or that does not go away. These are not all of the possible side effects of IBRANCE. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. | |||
How should I store IBRANCE?
Keep IBRANCE and all medicines out of the reach of children. | |||
General information about the safe and effective use of IBRANCE Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use IBRANCE for a condition for which it was not prescribed. Do not give IBRANCE to other people, even if they have the same symptoms you have. It may harm them. You can ask your pharmacist or healthcare provider for more information about IBRANCE that is written for health professionals. | |||
What are the ingredients in IBRANCE? Active ingredient: palbociclib Inactive ingredients: microcrystalline cellulose, colloidal silicon dioxide, crospovidone, magnesium stearate, succinic acid, HPMC 2910/hypromellose, titanium dioxide, triacetin, and FD&C Blue #2/Indigo Carmine Aluminum Lake. In addition, the 75 mg and 125 mg tablets contain red iron oxide and the 100 mg tablets contain yellow iron oxide.

For more information, go to www.pfizer.com or call 1-800-438-1985. | |||
Additional Resources
Chat online with Pfizer Medical Information regarding your inquiry on a Pfizer medicine or vaccine.
Speak with a Pfizer Medical Information Professional regarding your Pfizer medicine or vaccine inquiry.
Available 9AM-5PM ET Monday to Friday; excluding holidays.
Submit a medical question for a Pfizer medicine or a vaccine.
The submission will be reviewed during our standard business hours.
To report an adverse event related to a Pfizer product and you are not part of a clinical trial* for this medication, click the link below to submit your information:
Pfizer Safety Reporting Site
*If you are involved in a clinical trial for either product, adverse events should be reported to your coordinating study site.
If you cannot use the above website to report an adverse event related to a Pfizer medication, please call (800) 438-1985.
You may also contact the U.S. Food and Drug Administration (FDA) directly to report adverse events or product quality concerns either online at www.fda.gov/medwatch or by calling (800) 332-1088.